![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
119 Cards in this Set
- Front
- Back
|
Basic glycogen structure |
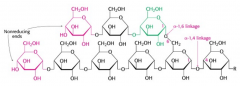
Includes non reducing ends and "reducing ends" on right
|
|
|
Glycogen breakdown (discovered by?) |
•Glycogen phosphorylase discovered by Carl and Gerty Cori •yields glucose 1-phosphate |
|
|
Two step mechanism of glycogen phosphorylase reacton |
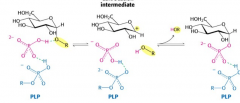
1.The phosphate, in conjunction with PLP (Pyridoal phosphate) Phophorylated vitamin B6, donates a hydrogen and releases the glycogen. (Makes schiff base) 2. The phosphate attacks the carbocation intermediate |
|
|
How glucose-1phosphate is converted to glucose 6 phosphate w/ mechanism |
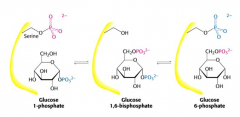
Phosphoglucomutase (generated by cleaving the 6 phosphate after phosphoglucokinase makes glucose 1,6 bisphosphate |
|
|
Glycogen breakdown in liver |
Glucose-6 phosphatase in membrane releases glucose into bloodstream |
|
|
Debranching in glycogen |
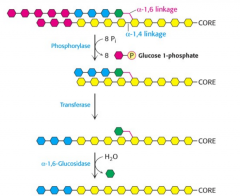
transferase and hydrolase in one polypeptide |
|
|
Glycogen synthesis |
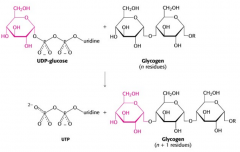
UDP-glucose is made by UDP glucose pyrophosphoylase, glycogen synthase gives glycogen +1 residues with retention of configuration |
|
|
Branching enzyme |
•Glucose alpha 1-4 linkages yields starch, not very soluble • Glycogen has alpha 1-6 branches and is more soluble •branching enzyme requires 11 or more glucose residues •removes about 7 residues and transfers them to alpha 16 linkage, more than 4 residues from the next branch |
|
|
Getting started with glycogen synthesis |
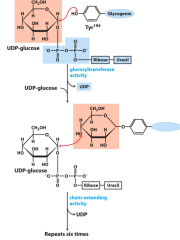
Glycogenin, ounce primer is made, glycogen synthase takes over |
|
|
Glycogen storage dieseases |
• PT had enlarged liver full of glycogen, low blood glucose between meals, Glucose-6-phosphatase is defective (Von Gierke) • PT's muscles cramp during vigorous exercise, blood lactate does not increase as much as in normal ppl --> defective muscle glycogen phosphorylase (McArdle), or defective PFK-1 (Tarui) |
|
|
Regulation of glycogen degradation |
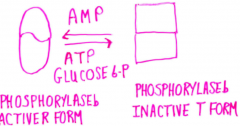
Allosteric control of muscle glycogen phosphorylase |
|
|
Hormone control of glycogen phosphorylase |
•Muscle: epinephrine •liver: glucagon •Epiniphrine or glucagon bind to receptors Receptor•hormone stimulates Gs-protein•GDP -->Gs-protein•GTP which stimulates adenyl cyclase, which makes cAMP PKA, which phosphorylates certain Sers (one such serine is on phosphylase-b kinase-SerOH |
|
|
Reversal of glycogen hormone cascade |

|
|
|
Regulation protein phosphatase 1 |
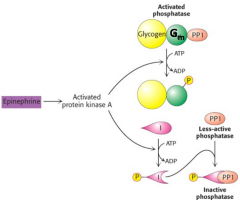
These phosphorylations must revert spontaneously |
|
|
Regulation of phosphorylase kinase by Ca++ release |
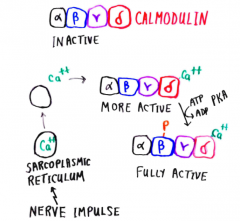
|
|
|
Glycogen phosphorylase of liver as a glucose sensor |
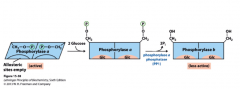
When glucose binds to allosteric sites, the phosphorylase becomes inactive (b) |
|
|
Epinepherine hormone cascade in heart (glycolysis and glycogenolysis) |

Glycogen synthase is turned off by the hormone cascade (phorphoylated), also phosphorylated by other kinases |
|
|
Coordinate control of glycogen metabolism |
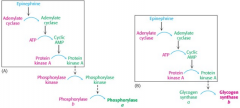
|
|
|
Citric acid cycle overview (discovery) |
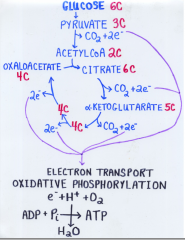
•O2 --> CO2 occurs in muscle but not cell extract •Centrifugation-> occurs in mitochondria |
|
|
Lipoic acid |
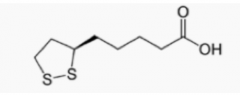
in amide linkage to a lysine side chain |
|
|
Citric acid cycle cofactors overview |
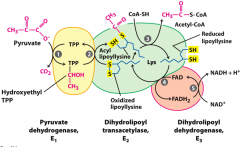
Pyruvate+ NAD+ CoA --> CO2 + acetyl-CoA _ NADH + H+ Cofactors: TPP, lipoate, NAD+, FAD, CoA |
|
|
Arsenic poisoning in citric acid cycle |
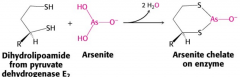
|
|
|
Citrate synthase reaction mech |
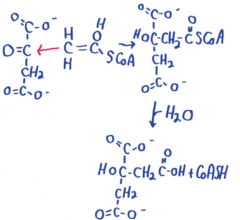
1 Acetyl-CoA (enol) + Oxaloacetate (with H2O) --> Citrate (with CoA-SH
|
|
|
Aconitase reaction |
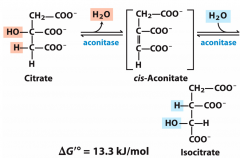
2 Iron sulfer cluster in aconitase pulls off OH- from C2 and transfers it to C3 |
|
|
Isocitrate dehydrogenase reaction |
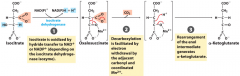
3 Loss of one CO2, coordinated by Mn2+ |
|
|
alpha-ketoglutarate dehydrogenase reaction |
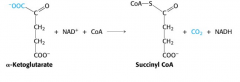
4 It is like pyruvate dehydrogenase reaction requiring NAD+, CoA, TPP, lipoate, FAD |
|
|
Conserved mechanism for oxidative decarboxylation |
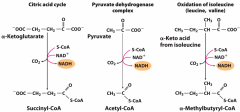
|
|
|
Succinyl-CoA synthetase reaction |
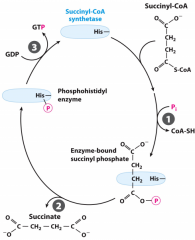
5 •converts succinyl-CoA to Succinate with GDP+ Pi to GTP and CoA-SH as a biproduct •GTP+ ADP --> GDP + ATP
|
|
|
Succinate dehyrdogenase reaction |
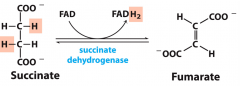
6 • Succinate --> Fumerate •FAD oxidized to FADH2 |
|
|
Fumerase reaction |
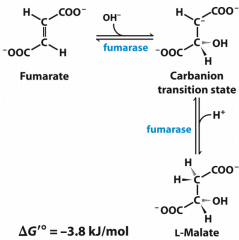
7 Has Carbanion transition state |
|
|
Malate dehydrogenase reaction |
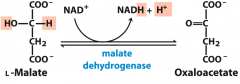
8 Oxidizes NAD+ to NADH + H+ |
|
|
Nine enzymes needed in citric acid cycle |
1 Citrate synthase 6 nucleoside 2 Aconitase diphosphate kinase 3 isocitrate 7 Succinate dehyrdogenase dehyrdogenase 4 a-ketoglutarate 8 Fumerase dehyrdogenase 9 malate 5 succinyl-CoA dehydrogenase
|
|
|
Comprehensive citric acid cycle |
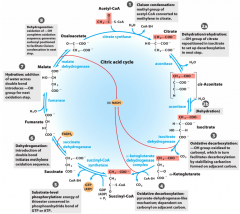
"officer can I keep selling sugar for money" = Oxaloacetate, citrate, isocitrate, ketoglutarate, succinyl-CoA, succinate, fumerate, malate |
|
|
An asymmetric enzme... |
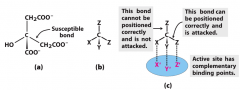
can bind a non-chiral compound asymetrically, they become chiral on the surface of the enzyme •compounds of this type are called prochiral |
|
|
Why reactions of NAD+ can get H on one side or the other |
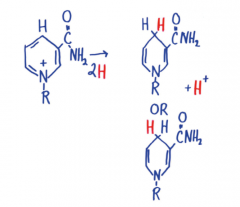
|
|
|
Why is oxidation of Acetyl-CoA a cycle |
•larger intermediates are easily bound and deformed by enzme surfaces, whereas acetate would be difficult to grab and manipulate. •Cycle allows the cell to make important molecules. -a-ketoglutarate --> Glutamate -oxaloacetate --> Aspartate
|
|
|
origin of oxaloacetate in TCA cycle |
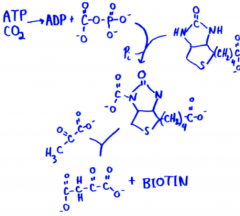
•produced during gluconeogenesis • allosteric activator is acetyl-CoA, (which accumulates from breakdown of fatty acids) |
|
|
Biotin |
A vitamin -requirement small for humans except for ppl who eat a lot of egg whites -avidin binds to biotin to prevent bacteria growth |
|
|
Reactions that fill up the citric acid cycle |
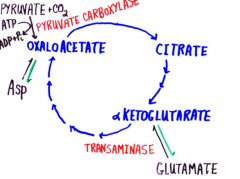
Anaplerotic |
|
|
Reactions drain the citric acid cycle |
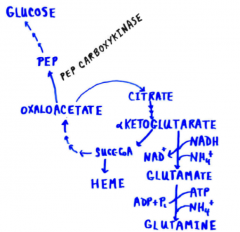
Cataplerotic |
|
|
Regulation of TCA |
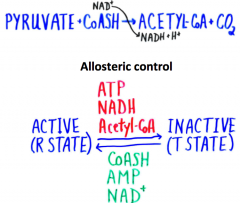
Pyruvate dehydrogenase reaction |
|
|
Covalent modification also regulates pyruvate dehyrogenase |

|
|
|
Regulation of isocitrate dehyrdogenase and communication between pathways |
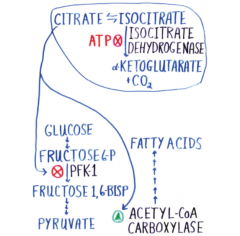
|
|
|
Reminder |
make notecards of involved systems to study afk |
|
|
Palmitate |
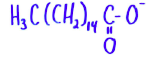
Hexadecanoic acid |
|
|
Glycerol |
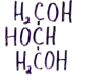
|
|
|
Triacyl glycerol |
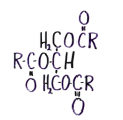
6 times better than carbs at storing energy, more reduced, not hydrated, GLY=2,500kJ, lasts for a day Fat = 420,000kJ lasts for months
|
|
|
Structure of adipocyte |
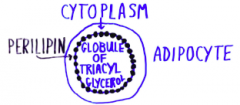
|
|
|
Release of fatty acids from triacylglycerols in the adipocyte |
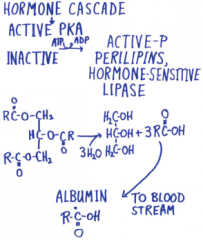
|
|
|
1904 Franz Knoop |
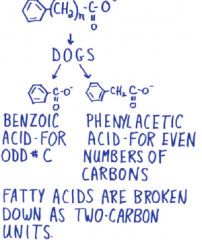
|
|
|
Fatty acid --> fatty acyl-CoA |
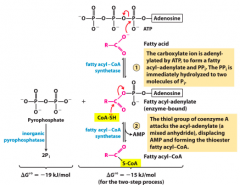
|
|
|
Carnitine |
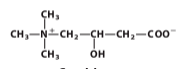
|
|
|
Entry of fatty acids into mitochondria |
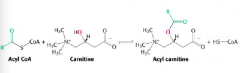
A carnitine acyl transferase reaction |
|
|
Acyl-carnitine/carnitine shuttle |
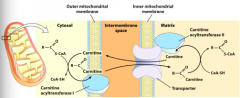
|
|
|
ß-oxidation scheme, part 1 |
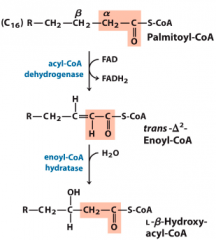
|
|
|
ß-oxidation scheme, part 2 |
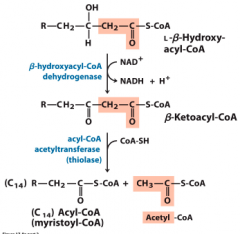
|
|
|
ß-oxidation scheme, part 3 |
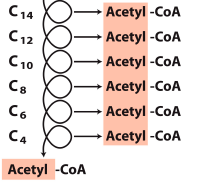
-Entry of electrons into electron transport and oxidative phosphorylation -FADH2--> electron transferring flavoprotein--> 1.5 ATP -NADH--> Complex I --> 2.5 ATP |
|
|
Regulation of fatty acid degradation and synthesis |
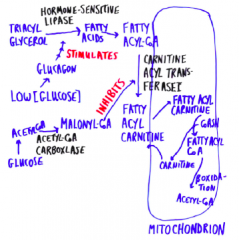
First step: ATP + CO2 + Acetyl-CoA carboylase --> ADP + Pi + Malonyl CA (inhibitor of carnitine acyl transferase I) ->->-> Fatty acids |
|
|
Diseases of fatty acid degradation (MCADD) |
Acyl-CoA dehydrogenases: 1 for long chains 12-18 1 for medium chains 2-14 1 for short chains 4-8 - Medium Chain AD deficiency, MCADD, Lys304Glu--> 10%SIDS --> high levels of octanoic acid in blood, dicarboxylic acids in urine Tx: low fat, high carb diet |
|
|
Diseases of fatty acid degradation (Jamaican vomiting sickness) |
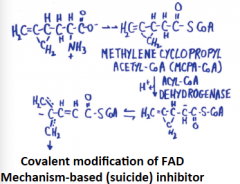
Convulsions, coma, and death caused by Unripe Aackee fruit that contains hypoglycin |
|
|
Degradation of fatty acids with odd numbers of carbons |
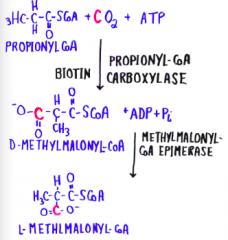
|
|
|
Vitamin B-12 |
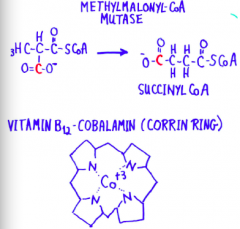
|
|
|
Arrangement at the rxn center of Vitamin B12 |
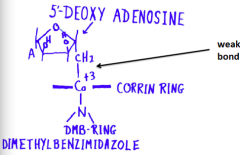
|
|
|
Mechanism of C and H rearrangements (B12) |
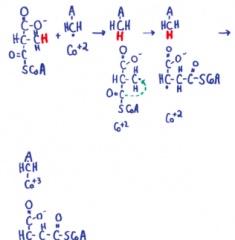
|
|
|
Vitamin B12 disease |
Vitamin B12 is made by bacteria and used by people Plants do not use B12 because it is photosensitive People absorb B12 through intenstine by a protein called intrinsic factor, geriatrics are deficient in intrinsic factor --> get pernicious anemia, need B12 injections |
|
|
Intrinsic factor found by? |
1918 Hooper: Raw liver cures pernicious anemia in PTs 1925 Whipple: induced anemia in dogs, cured with diet of raw liver 1926 Minot and Murphy: showed that eating 1/2 lb of raw liver /day cured pernicous anemia 1936 Castle: PTs w/ Stomachs removed --> pernicious anemia, vomitus cured PTs |
|
|
Extrinsic factor |
1947 Shorb & Flokers: Factor purified from liver allows growth of bacterium on minimal salts and glucose 1956 Hodgkin: Extrinsic factor = Vitamin B12
Intrinsic factor ID'd w/ radioactive Cobalt, binds to Co, Vit B12 used by humans in methylmalonyl-Coa Mutase and methionine synthase |
|
|
Glycerol into glycolysis |
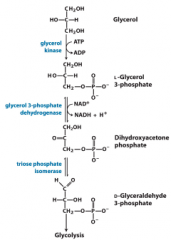
|
|
|
Starvation, diabetes --> |
![Accumulation of "ketone bodies"
In liver there is high [acetyl-CoA] and low [oxaloacetate]](https://images.cram.com/images/upload-flashcards/13/46/83/7134683_m.png)
Accumulation of "ketone bodies" In liver there is high [acetyl-CoA] and low [oxaloacetate] |
|
|
origin of ketone bodies (1) |
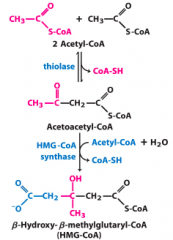
|
|
|
Origin of ketone bodies 2 |
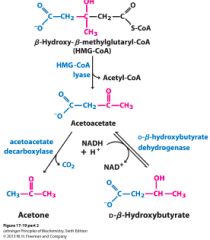
ß-hydroxybutyrate travells to the brain |
|
|
ß-hydroxybutyrate as fuel in TCA cycle |
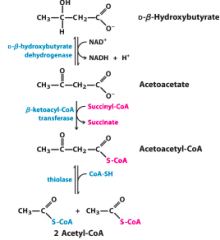
Oxaloacetate may come from protein breakdown aspartate--> oxaloacetate |
|
|
High levels of ß-hydroxybutyrate reduce blood pH (Acidosis) |
Extreme acidosis is called ketosis which can cause coma and death, if dieting, eat some carbs to provide oxaloacetate |
|
|
Essential AAs |
V,I,L(branched chain) F, W (aromatic) T, H, K, M
Amino acids cannot be stored Excess amino acids --> keto acids and are used as fuel |
|
|
AA digestion methods |
Stomach: pepsin /F/Y/W Small intestine: trypsin K/R/ Chymotrypsin F/W/Y/ Carboxypeptidase releases AA from C-terminus Aminopeptidase releases AAs from the N-terminase |
|
|
Enzyme catalyzed transaminations |
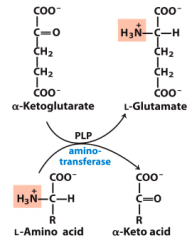
|
|
|
Transamination mech pt. 1 |
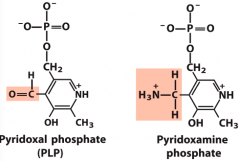
|
|
|
Transamination mechanism pt. 2 |
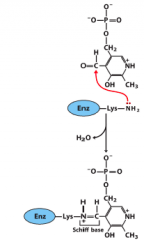
Includes nitrogen attack and schiff base |
|
|
Transamination mechanism pt. 3 |
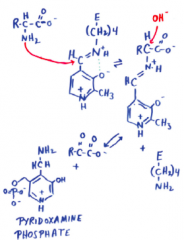
|
|
|
Glutamate dehyrdogenase reaction |
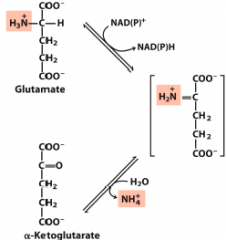
Glu+NAD(P)+ + H20--> alpha-ketoglutarate + NADPH + NH4+, Driven by NH4 removal |
|
|
Delivery of amino groups to the liver for the urea cycle |
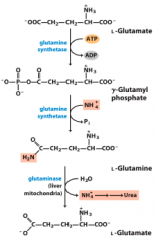
|
|
|
Delivery of alanine to liver |
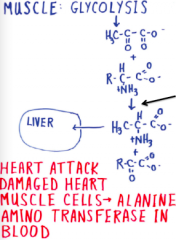
alanine aminotransferase |
|
|
Urea synthesis in liver stimulated by? |
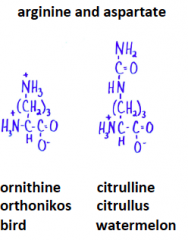
Stimulted 10-fold in excess of molar amount of certain added compounds |
|
|
Overall scheme of urea cycle |
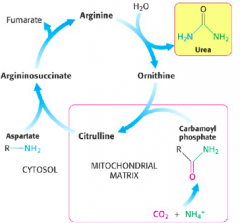
|
|
|
Reactions of urea cycle |
-Carbomoyl phosphate synthetase I -Ornithine transcarbamoylase -Argininosuccinate -Argininosuccinase, arginase |
|
|
Carbamoyl phosphate synthetase I |
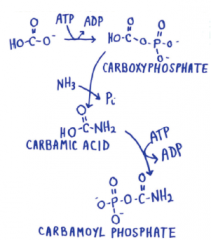
|
|
|
Ornithine transcarbamoylase |
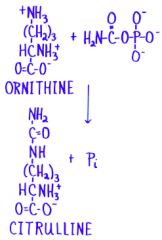
|
|
|
Arginosuccinate synthetase |
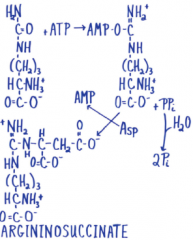
|
|
|
Argininosuccinase |
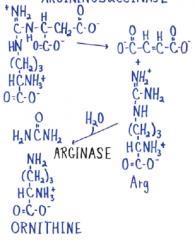
|
|
|
Keto-amino cycle |
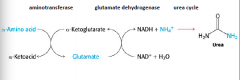
|
|
|
Carbamoyl phosphate synthetase I deficiency |
![leads to high [NH4+] in blood. Tx: Low protein diet, alternative nitrogen excretion](https://images.cram.com/images/upload-flashcards/15/38/98/7153898_m.png)
leads to high [NH4+] in blood. Tx: Low protein diet, alternative nitrogen excretion |
|
|
Argininosuccinate Acidemia |
argininosuccinase deficiency, argininosuccinate accumulates and is excreted Arginine is depleted Tx: Low protein diet + arginine |
|
|
Big picture of urinary system pt. 1 |
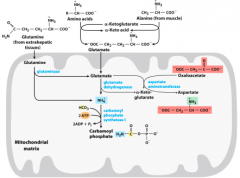
|
|
|
Big picture of urinary system pt. 2 |
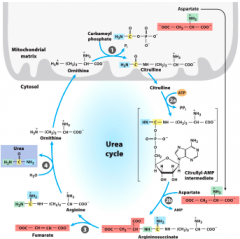
|
|
|
Diseases in converting breakdown of AA |
Alkaptonuria- black urine (recessive traits) Homogentisate accumulates, gives arthritis late in life (know mechanism?) Missing Homogentisate dioxygenase Phenylketonuria- Urine+ Fe3+ --> olive green, PTs have high levels phenylpyruvate Tx: Low Phe diet, no aspartame (D-F-methyl-ester) Phenylalanine is defective in PKU, Phe competes with other AAs in nerve cells
|
|
|
Branched chain AAs pt 1 |
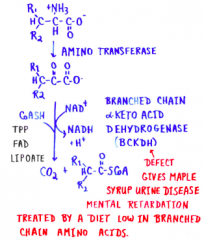
|
|
|
Branched chain AAs pt 2 |
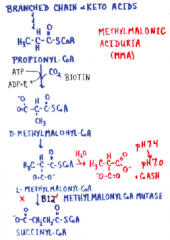
|
|
|
Patricia Stallings case |
MMA responsible for poisoning Tx: diet low in fatty acids with odd numbers of carbons, branched chain amino acids, and methionine |
|
|
Methionine metabolism |
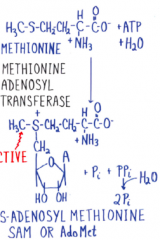
|
|
|
SAM donates methyl groups |
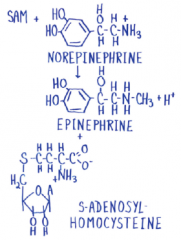
Can also donate methyls to DNA protecting it |
|
|
Regeneration of SAM |
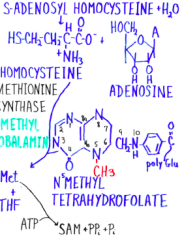
|
|
|
Degradation of methionine contributes to MMA |
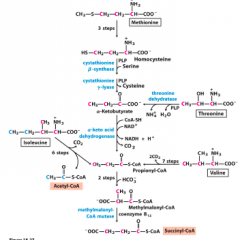
|
|
|
Ox Phos Cytochrome peaks |
c types: 550nm b types: 560nm a types: 600nm |
|
|
order of reduction of cytochromes |
cyt b, cyt c1 cytc, cyt a, O2 |
|
|
Coenzyme Q mechanism |
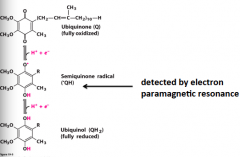
|
|
|
Last discovered electron carriers |
Iron-sulfur centers, Oxidation states detected by EPR |
|
|
COMPLEX I |
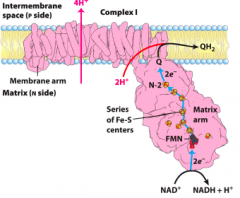
NADH: ubiquinone oxidoreductase NADH+5H+N+Q>NAD+ +QH2+4H+P |
|
|
COMPLEX II (succinate dehydrogenase) |
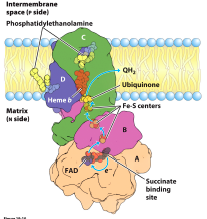
No H+ is pumped across the inner membrane. No ATP is synthesized. Mutations in Heme b allow more Q- to donate electrons to •O2 --> superoxide radical--> tumors of the head and neck (hereditary paraganglioma) |
|
|
Fatty-acyl-CoA dehydrogenase |
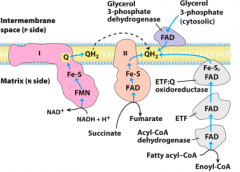
No pumping of H+ No ATP |
|
|
COMPLEX III (Cytochrome bc complex = Ubiquinone: cytochrome c oxidoreductase) |
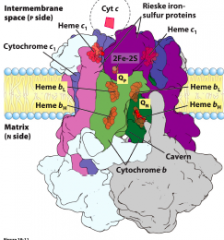
Electrons will pass from QH2, a two-electron carrier to cyt c, a one-electron carrier |
|
|
Complex III and the Q cycle |
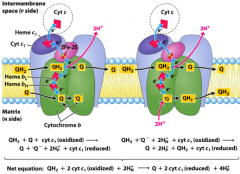
|
|
|
Complex IV, Cytochrome c oxidase |
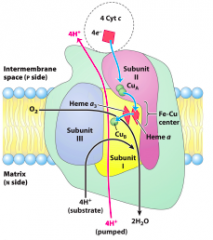
4 cyt c (red) = 8 H+(N) + O2 --> 4 cyt c (ox) + 4H+(p) + 2H2O |
|
|
F fragment ATPase |
F_1 (upper) has 3 equivalent has 3 equivalent alpha-beta units and it makes ATP F_0 (oligomycin-sensitive, lowers) pumps H+ and causes the asymmetric gamma center stalk to turn. The alpha-beta subunits of F_1 bind to ATP or ADP + Pi, or nothing |
|
|
Symporter for Pi and protons |
membrane protein that transports the species in the same direction |
|
|
Poisons for electron transport and ATP synthesis |
Rotenone (complex I) antimycin A (complex III) cyanide (complex IV) oligomycin B (F-ATPase) bongkerekic acid/ atracytlocide for adenine nucleotide translocase |
|
|
Genetic diseases of mitochondria |
hereditary paraganglioma Leber's hereditary optical neuropathy (LHON) myoclonic epilepsy and ragged red fiber disease (MERRF)
Creation and destruction of superoxide radicals |
|
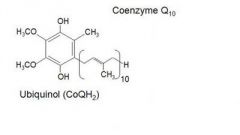
|
ubiquinone (coenzyme Q) |
|
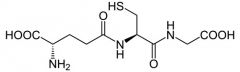
|
Glutathione (gamma-glutamyl-cysteinyl-glycine) |
|
|
2,4 dinitrophenol |
Do you know how to draw this? |