Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
95 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Pyruvate kinase deficiency
|
Glycolytic enzyme deficiency
Pres: Hemolytic anemia d/t inability to maintain Na/K- ATPase --> RBC swelling/lysis. Also caused by phosphoglycose isomerase deficiency. |
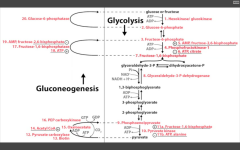
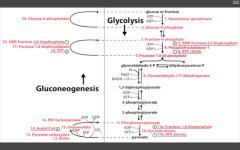
Glycolysis
|
|
|
A muscle biopsy reveals elevated glycogen, elevated F6P, and decreased pyruvate. What is the deficient ENZ?
|
PFK1
Most likely ENZ deficiency because the substrate just before has accumulated. |
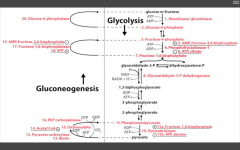
Most likely.
|
|
|
RLS of glycolysis?
|
PFK-1
Stimulated by AMP (need more ATP) and F26BP (fed state indicative via stim of PFK-2 by insulin) Inhibited by ATP (enough ATP) and Citrate (product of TCA cycle) |
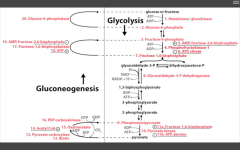
Catalyzes F6P --> F16BP step, just before F16BP is split to GA3P and Dihydroxyacetone-P
|
|
|
RLS of Gluconeogenesis?
|
F16-bisphosphatase
Accomplished in liver. Stimulated by ATP (enough ATP, need to store Energy/share it with other cells) Inhibited by AMP (need more ATP), and F26-BP (fed state, should make E) - molecules of E deficiency. Requires Mg2+ to function. |
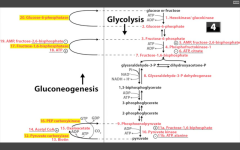
Catalyzes F16BP-->F6P step in the liver of gluconeogenesis, and in the Calvin cycle (reverse of PFK-1 in glycolysis).
|
|
|
RLS of TCA (Krebs) cycle?
|
Isocitrate Dehydrogenase
|
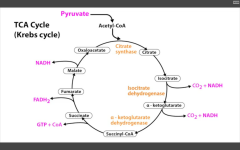
Catalyzes Isocitrate --> alpha-KG (yielding CO2 + 1NADH).
|
|
|
RLS of Glycogen synthesis?
|
Glycogen synthase
NOTE: glucose--> G6P--> G1P in glycogen synthesis!! Also, UDP-glucose pyrophosphorylase primes the glucose to be attached to the growing glycogen chain. Attached to the molecule by glycogen synthase (makes alpha-1,4 bonds. Branching enzyme makes branches (and alpha-1,6 linkages). Skeletal muscle and Hepatocytes. |
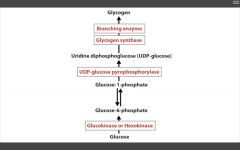
Catalyzes UDP-glucose --> Glycogen, along with Branching ENZ
|
|
|
RLS of glycogenolysis?
|
Glycogen phosphorylase
inhibited by ATP (enough!), and glucose (why break it down when you've already got it goin'?) Alpha-1,6 glucosidase AKA de-branching ENZ - breaks it down directly into glucose. |
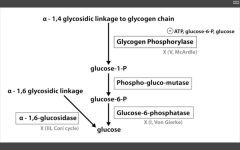
Catalyzes alpha-1,4 glycosidic linkage break to release G1P.
Glucose-6-phosphatase is absent in muscles. This is the last step to gen glucose for gluconeogenesis and glycolysis...this is why muscles are so selfish. |
|
|
RLS of HMP shunt?
|
G6P-DH
|
Catalyzes G6P --> 6-phosphoglyconolactone to generate more NADPH and pentose sugars.
Function of PPP: Generate reducing equiv (NADPH) [60% of total NADPH production] Produce R5P (used to synth nucleotides/nucleic acids Produce E4P used to synth aromatic AA Digest dietary pentose sugars to form glycolytic intermediates Most active in liver, mammary, and adrenal gland |
|
|
RLS of de-novo pyramidine synthesis?
|
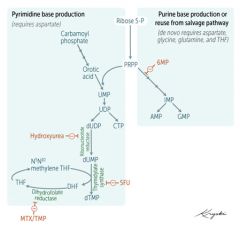
CPS-II
carbamoyl phosphate II |

catalyzes conversion of ATP, CO2, and glutamine to Carbamoyl-P, which is then coverted to base (orotic acid).
|
|
|
RLS of de novo purine synthesis?
|
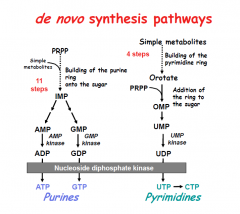
PRPP-amidotransferase
|
HINT: PUR As Gold.
catalyzes PRPP --> phosphoribosyl amine. |
|
|
Purine salvage abnormalities?
|
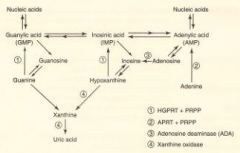
HGPRT - lesch-nyhan
XLR Adenosine deaminase deficiency Auto recessive |
HGPRT - catalyzes guanine-->GMP AND hypoxanthine -->IMP.
Defective purine salvage results in XS uric acid production and de novo purine synthesis. Hint: He's Got Purine Recovery Trouble. Adenosine deaminase def: catalyzes adenosine-->Inosine. Excess ATP and dATP imbalances nucleotide pool via feedback inhibition of ribonucleotide reductase. Prevents DNA synthesis and decreases lymphocyte count. A major cause of SCID. Hint: SCID happens to Kids. |
|
|
Malate aspartate shuttle
|
Heart, liver
|
Gen 32 ATP per molecule
|
|
|
G3P shuttle
|
Muscles
|
Generate 30 ATP, less efficient
|
|
|
GLUT-1
|
RBC, brain - insulin-independent
|
Only E metabolite they recognize
|
|
|
GLUT-2
|
hepatocytes, beta cells of pancreas
|
|
|
|
GLUT-4
|
insulin-dependent
skeletal muscle, adipose tissue |
only when you need to bring Glucose in according to systemic levels
|
|
|
Hexokinase vs. Glucokinase
|
Glucokinase - high Km, high Vmax
beta cells, hepatocytes Hexokinase - low Km (high affinity for glucose), low vMax not induced by insulin! |
difference between cells that regulate glucose metabolism vs. those that need glucose less conditionally.
|
|
|
Regulation of Pyruvate kinase
|
Stim by F16BP (upstream substrate)
Inhib by ATP and alanine (indicates that you have plenty of E) |
Catalyzes PEP --> pyruvate (last step before entering TCA cycle.
|
|
|
Regulation of intracellular F26BP levels?
|

PFK-2 increases levels of F26BP
FBP2 decreases levles of F26BP |
PFK2 is stimulated by insulin in FED STATE. Converts F6P to F26BP (take it away from gluconeogenesis). F26BP stimulates PFK1 to convert F6P to F16BP --> glycolysis.
FBP2 is stimulated in FASTING STATE, and converts F26BP to F6P (enter into gluconeogenesis). |
|
|
Thiamine in TCA cycle
|
Vitamin B1
These dehydrogenase reactions generate NADH in the mitochondria which enter the electron transport chain to generate ATP, therefore the patient has a problem making ATP. Cofactors required by the above 2 enzymes are Thiamine Lipoic acid CO2 Folate NAD+ Which can be remembered by the phrase, "Tender Love and Care for Nancy" |
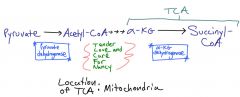
Oxidative decarboxylation in TCA cycle
|
|
|
Epinephrine's effect on glycolysis (specifically, its effect on liver, pancreas, and muscles).
|
1. Liver: Stimulates Beta-2 receptors --> glycogen breakdown.
2. Pancreas: Stimulates glucagon release (+ adenylate cyclase --> increased cAMP --> more active FBP2 --> converts F2,6BP --> F6P; less PFK2 active --> less activation of PFK1 --> less activation of glycolysis. 3. Muscles: + beta-2 receptors to break down glycogen to glycose. Muscle doesn't respond to glucagon, so F26BP increases --> activates PFK1 --> more glycolysis. SUGAR WHERE YOU NEED IT. |
Hint - think of whatchu gotta do when you fightin' or flightin'
|
|
|
What cofactor does Pyruvate carboxylase require?
|
Biotin
Pyruvate carboxylase catalyzes conversion of pyruvate directly to OAA. Stimulated by Acetyl co-A (plenty of E around, so need to go back up pathway). |
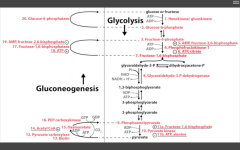
Carboxylation requires biotin.
|
|
|
Key steps of Gluconeogenesis?
|
Key ENZ highlighted:
Pyruvate carboxylase (+biotin) PEP carboxylase F16BPase G6Pase Occurs in cells designed to share glucose: Liver hepatocytes, kidney, intestinal epithelium Happens because you don't have much glucose in your blood. Liver shares glycogen stores, and forms pyruvate to go into glycolysis. |
Muscles don't have glucose 6 phosphatase
|
|
|
Why can't muscles undergo gluconeogenesis?
|
They don't have glucose-6 phosphatase (required to dephosphorylate G6P to yield glucose).
|
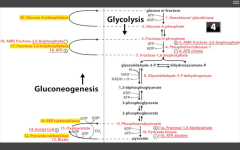
Yoooo
|
|
|
Which pathways lead to gluconeogenesis?
|
1. Odd chain FAs --> propionyl coA --> pyruvate --> gluconeogenesis
2. Krebs cycle molecules --> gluconeogenesis 3. Molecules --> OAA --> gluconeogenesis 4. AA --> TCA substrate, OAA, pyruvate |
Many things can enter into the gluconeogenesis pathway.
|
|
|
Can muscles generate glucose from glycogenolysis?
|
No, they lack the ENZ glucose-6-phosphatase (the same ENZ required to complete gluconeogenesis), so the G6P they generate directly enters glycolysis.
|
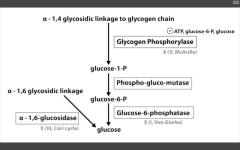
Muscles never share Energy!
|
|
|
Glycogen storage diseases: Name them and tell me why! (x4)
|
1. Von Gierke's disease (Type 1)
- G6 Phosphatase deficiency. Can't get free glucose from cell, can't share glucose from liver, kidneys. Severe fasting hypoglycemia, TONS of glycogen in liver, increased blood lactate levels (cells are starved for energy), increased uric acid (lactic acid/uric acid compete for excretion), and hepatomegaly (cells are swollen). 2. Pompe's disease (Type II) - deficiency of lysosomal alpha1,4 glucosidase Can't break alpha-1,4 glycosidic bonds. Infantile and adult forms. (deets on hint) 3. Cori's disease (Type III) - deficiency of debranching ENZ (alpha-1,6 glucosidase) milder Type I (von Gierke's) with normal blood lactate levels. 4. McArdle's disease (Type V) - deficiency of skeletal muscle glycogen phosphorylase. Glycogen accumulates in muscle, but can't break it down. Draws in H20, cells swell and some lyse. Painful muscle cramps, myoglobinuria with strenuous exercise. (myoglobin in serum --> myoglobinuria --> clogs kidneys and causes Rhabdomyolysis). |
Very Poor CHO Metabolism
Von Gierke's: Auto recessive. Pompe's trashes the Pump. Auto recessive. Cori's: Auto recessive. Gluconeogenesis is intact. McArdle's: Auto recessive. McArdle's = Muscles. |
|
|
Glycogen phosphorylase deficiency
|
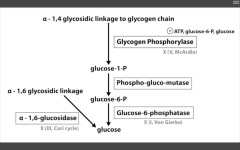
McArdle's (V)
|
|
|
|
Glucose-6-phosphatase deficiency
|
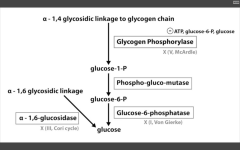
Von Gierke's disease (I)
|
|
|
|
Lactic acidosis, hyperlipidemia, hyperuricemia (Gout)
|
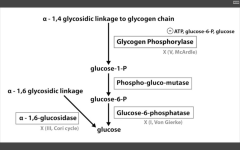
Von Gierke's disease (I)
|
|
|
|
Alpha 1,6 glucosidase deficiency
|
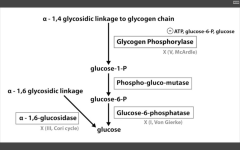
Cori's disease (III)
|
|
|
|
alpha 1,4 glucosidase deficiency
|
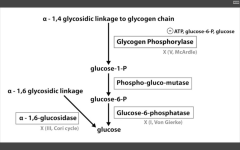
Pompe's disease (II)
|
Very Poor CHO Metabolism
|
|
|
Cardiomegaly (in a glycogen storage disorder)
|
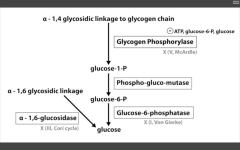
Pompe's (II) - infantile variant (early death)
|
Pompe's trashes the Pump (heart, liver, muscle)
Very Poor CHO Metabolism |
|
|
Diaphragm weakness leading to respiratory failure (in a glycogen storage disease)
|
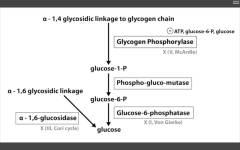
Pompe's (II) - Adult variant
|
Pompe's trashes the pump (heart, liver, muscle)
Very Poor CHO Metabolism |
|
|
Increased glycogen in liver, severe fasting hypoglycemia (glycogen storage disease)
|
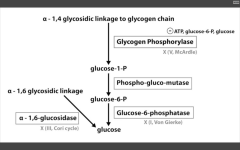
Von Gierke's (I)
|
|
|
|
Hepatomegaly, hypoglycemia, hyperlipidemia (normal kidneys, lactic, and uric acid)
|
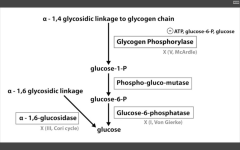
Cori's disease (III)
|
|
|
|
Painful muscle cramps, myoglobinuria with strenuous exercise
|
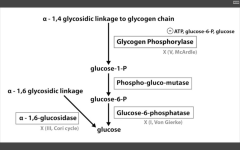
McArdle's (V)
|
|
|
|
Severe HSM, enlarged kidneys
|
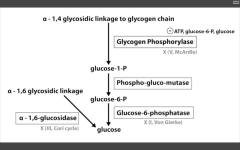
Von Gierke's disease (I)
|
|
|
|
Co-factors required for PDH (pyruvate DH) complex:
|
1. Thiamine (B1) --> TPP
2. Lipoic acid 3. CoA (B5, panthothenate) 4. FAD (B2, riboflavin) 5. NAD (B3, Niacin) |
Tender Loving Care For No one
|
|
|
Fates of pyruvate:
|
1. Oxaloacetate via Pyruvate carboxylase (to TCA cycle or gluconeogenesis)
2. Acetyl co-A via PDH (pyruvate DH) from glycolysis to TCA cycle 3. Lactate via Lactic acid DH to anaerobic glycolysis Cori cycle (RBC, leukocytes, kidney medulla, lens, testes, cornea) 4. Alanine via Alanine aminotransferase (B6) to carry amino groups to liver. |
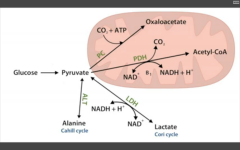
APPL
|
|
|
Which enzyme does Arsenic interfere with? What are the sx of exposure?
|
Arsenic inhibits lipoic acid, which interferes with the functioning of Pyruvate DH complex.
Sx: vomiting, rice-water stools, and garlic breath. Looks like gastroenteritis with garlic breath. |
|
|
|
Key enzymes in TCA cycle:
|
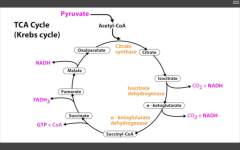
1. Citrate synthase
2. Isocitrate DH 3. alpha-KG DH Produces 3NADH, 1FADH2, 2CO2, 1GTP per acetyl-coA = 10ATP/acetyl coA = 20ATP/glucose. |
Occurs in mitochondria.
|
|
|
Cofactors required for alpha-KG DH complex:
|
Same cofactors as for pyruvate DH complex of last committing step of glycolysis:
Thiamine (B1) --> TPP Lipoic acid CoA (B5, pantothenate) FAD (B2, riboflavin) NAD (B3, niacin) |
Tender Loving Care For No one
Arsenic inhibits lipoic acid. |
|
|
Pyruvate DH deficiency:
|
causes backup of substrate (pyruvate and alanine) --> lactic acidosis.
Causes: 1. X linked mut of E1-alpha subunit of PDHC 2. Acquired (arsenic exposure, Vit B1, B2, B3, B5 deficiency). Findings: Neurologic deficits, usually starting in infancy. Rx: increase intake of ketogenic nutrients (high fat content OR high lysine/leucine) |
Lysine and Leucine are the onLy pureLy ketogenic amino acids.
|
|
|
Describe the 5 complexes of the ETC?
|
Complex 1 - accepts electrons from NADH, moves H+.
Complex 2 (succinate DH) - accepts electrons from FADH2, moves H+. Passes electrons to CoQ. Complex 3 - receives electrons from Complexes 1 and 2. Passes them via cytochrome c to Complex 4. Complex 4 - passes electrons to final electron acceptor, O2 --> H2O. Moves H+. Complex V - ATPase --> makes ATP using the energy from H+ gradient. |
|
|
|
Name the electron transport inhibitors:
|
Complex 1 - inhibited by Rotenone, Amytal (barbituate) and MTP (from MPTP, which is what you make by accident if you're trying to home brew demerol or riperidine). [electron transport inhibitor]
Complex 2 - Nothing. Complex 3 - Antimycin A [electron transport inhibitor] Complex 4 - CN, CO, N3- (sodium azide), and H2S (hydrogen sulfide). [electron transport inhibitors] Directly inhibit electron transport, causing decreased proton gradient and blocked ATP synthesis. |
RAM A CONCH, OH!
|
|
|
Name the ATP synthase inhibitor:
|

Complex 5 - Oligomycin (ATP synthase inhibitor).
Directly inhibit ATPase, causing increased H+ gradient. No ATP produced because electron transport stops. |
RAM A CONCH, OH!
|
|
|
Name the uncoupling agents:
|
2,4-DNP, ASA, thermogenin.
increase permeability of membrane, causing decreased proton gradient and increased O2 consumption. ATP synthesis stops, but electron transport continues. |
DaT ASS is full of (brown) fat!
|
|
|
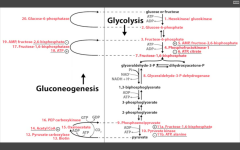
Name the irreversible ENZ in gluconeogenesis:
|
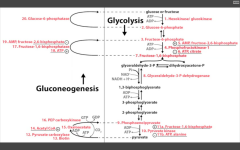
1. Pyruvate carboxylase (pyruvate--> OAA)
---requires biotin, ATP. ---activated by acetyl coA 2. PEP carboxykinase (OAA --> PEP) ---requires GTP 3. F16BPase (F16BP-->F6P) G6Pase (G6P --> glucose) |
Pathway Produces Fresh Glucose
|
|
|
What is the deficiency in essential fructosuria?
|
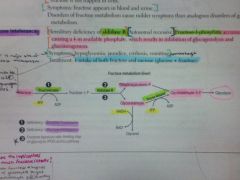
Fructokinase
Benign, as fructose isn't trapped in cells. Sx: fructose appears in blood and urine. |
Fructose appears in urine/blood.
|
|
|
What is the deficiency in Fructose intolerance?
|
Aldolase B
F1P accumulates, causing a decrease in available phosphate. Inhibits gluconeogenesis and glycogenolysis. Sx: hypoglycemia, jaundice, cirrhosis, vomiting, hemorrhage. rx: decrease intake of both fructose and sucrose (glucose+fructose). |
F1P accumulates in cells.
FAB GUT. |
|
|
What results from a deficiency of galactokinase?
|
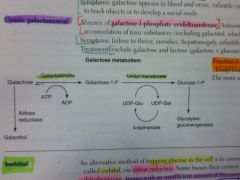
Galactokinase deficiency.
Galactitol accumulates if galactose is present in diet. Mild. Sx: galactose in urine/blood, infantile cataracts. May initially present as failure to track objects or failure to develop a social smile. |
Accumulates in lenses.
|
|
|
What results from a deficiency of galactose-1P uridyl transferase?
|
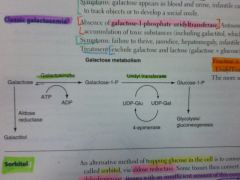
Classic galactosemia.
Auto recessive. Accumulation of toxic substances (like galactitol), which accumulates in lens of eye. Sx: failure to thrive, jaundice, hepatomegaly, infantile cataracts, MR (builds up in neural tissues). |
FAB GUT
Fructose is to Aldolase B as Galactose is to UridylTransferase |
|
|
Which are the important enzymes in the oxidative burst pathway?
|
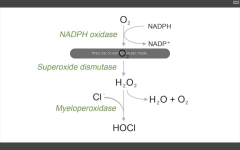
1. NADPH oxidase
(O2-->O2-.) 2. Superoxide dismutase (O2-. --> H2O2) 3. Myeloperoxidase (H2O2 +Cl --> HOCl.) |
|
|
|
What is the defiency in CGD?
|
Chronic granulomatous disease:
NADPH deficiency (can't convert O2-->O2-.) Phagolysosomes can't make caustic substances to digest bact. Macrophages can make H2O2 from Env and use that to digest bacteria. Bacteria with catalse can digest that H2O2, so phagocytes can't use it to digest bacteria. |
|
|
|
Draw the Lipid transport system:
|
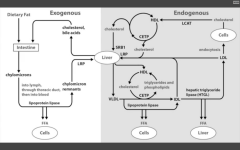
Note the deficiencies.
|
|
|
|
Name the essential amino acids:
|
Phenylalanine
Valine Threonine Tryptophan Isoleucine Methionine Histidine Leucine Lysine |
PVT TIM HaLL
|
|
|
Name the acidic amino acidS:
|
Aspartate and Glutamate (aspartic acid and glutamic acid)
|
GA! acid!
|
|
|
Name the basic Amino Acids:
|
Arginine, Lysine, Histidine
Arginine is most basic Histidine has no charge at body pH |
HAL is so basic.
|
|
|
Which two amino acids are required during times of growth?
|
Arg and His
|
Growing up, hahaha
|
|
|
Which two amino acids are in high concentrations in histones?
|
Arg and Lys
|
DNA's song - lalalalala
|
|
|
AA derivatives of Phenylalanine?
|
--> tyrosine (+BH4) via phenylalanine hydroxylase (missing in PKU)
--> DA (+BH4) via DA hydroxylase - inhibited by carbidopa --> Dopamine (+Vit B6) via DA decarboxylase - breakdown product: HVA --> NE (+Vit C) - Breakdown product: VMA --> Epi (+SAM) - ramped up by cortisol - Breakdown product: Metanephrine |
Pheeny's That Damn Dope who Needs Everything.
|
|
|
AA derivatives of Tryptophan?
|
Niacin (+B6) --> NAD+/NADH
Serotonin (+BH4) --> Melatonin (pineal gland) |
Tripping NiSe....energy and then sleep
|
|
|
AA derivatives of Histidine?
|
Histamine (+B6)
|
HH Gregg.
|
|
|
AA derivatives of Glycine?
|
-->porphyrin (+B6)
--> Heme (RLS aminolevulinic synthase) |
Glycines pH
|
|
|
AA derivatives of Glutamate?
|
--> GABA (B6) - inhibitory NT
--> Glutathione - reducing agent |
Glutes - All Gab, no gluttony.
|
|
|
AA derivatives of Arginine?
|
--> Creatitine (high E Phosphate carrier to supply ADP)
--> Urea --> NO (vasodilator/smooth muscle relaxer) |
Arghhh, ye mateys!! Create ur NOses now!
|
|
|
Which is the most common type of urea cycle enzyme deficiency?
|
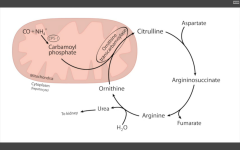
Ornithine transcarbamoylase
Sx. of hyperammonemia: - slurring speech somnolence vomiting cerebral edema blurred vision (aka hepatoencephalopathy) XLR xs carbamoyl phosphate --> orotic acid Findings: increased orotic acid in urine, DECREASED BUN (can't release nitrogen), and sx of hyperammonemia. |
|
|
|
What is the RLS of the urea cycle?
|
CPS1 - converts NH4+ CO2 --> carbamoyl phosphate, using 2ATP
|
|
|
|
Sx of hepatoencepalopathy, and how it arises?
|
SX:
Slurred speech, somnolence, vomiting, cerebral edema, blurred vision Arises: acquired (liver dz) hereditary (urea cycle ENZ deficiencies, e.g. ornithine transcarbamoylase def) |
If you've got too much ammonia, you're slurring your vomit while sleepy, with blurry vision and a h/a.
|
|
|
Rx for hepatoencephalopathy?
|
Bind AA so it can be excreted:
Phenylbutyrate Benzoate Acidify GI tract/trap NH4+ for excretion: Lactulose |
Trap PB, Acid milk.
|
|
|
Deficiency in PKU?
|
Decreased phenylalanine hydroxylase OR BH4 (Phe --> tyr), so tyrosine becomes essential. Increased [phenylalanine] --> phenylketones in urine.
Phenylketones: phenylacetate, phenyllactate, phenylpyruvate. Findings: MR, growth retardation, seizures, fair skin, eczema, musty body odor. Rx: decrease Phe, supplement Tyrosine or BH4 within 2-3 wks of birth. AR, 1/10,000. Screen 2-3 days after birth b/c maternal tyrosine has worn off and phenylketones are now detectable. |
Disorder of Aromatic AA metabolism --> Musty BO
|
|
|
Deficiency in Alkaptonuria?
|
Homogentisic acid oxidase (degradative pathway of tyrosine to fumarate.
AR, benign. Findings: dark CT, brown pigmented sclera, urine tyrnes black on prolonged exposure to air. May have debilitating arthralgias. Presents in adulthood. |
|
|
|
Deficiency in Albinism?
|
1. Tyrosinase (inability to synth melanin from tyrosine)
2. Defective tyrosine transporters (decr amt of tyrosine, thus decr melanin) 3. lack of migration of neural crest (melanocytes). increased risk of skin cancer, variable inheritance. |
|
|
|
Deficiency in homocystinuria?
|
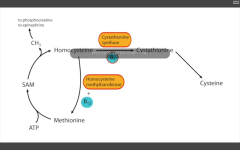
1. cystathionine enthuse (rx by decreasing methionine and cystine, and increasing B12 and folate in diet)
2. decreased affinity of cystathione synthase for pyridoxal phosphate (rx increase amt B6 in diet) 3. homocysteine methyltransferase (requires B12) def all result in xs homocysteine, and cysteine becomes essential. Findings: increased homocystein in urine, MR, osteoporosis, tall stature, kyphosis, lens subluxation, and atherosclerosis. |
|
|
|
Deficiency in cystinuria?
|
Defect in COLA transporter
- Cysteine - Ornithine - Lysine - Arginine PCT of the nephron. Xs cystine --> precipitation of hexagonal crystals and renal staghorn calculi AR, common (1,7000) Rx: hydration and urinary alkalinization (Acetazolamide) - carbonic anhydrase inhibitor, alkalinizes urine to prevent stones. |
cystine COLA
|
|
|
Deficiency in maple syrup urine disease?
|
Blocked degradation of BRANCHED AA (Ile, Leu, Val) d/t decreased branched alpha ketoacid DH (B1) complexes. Causes increased alpha keto acids in urine, esp Leucine.
Pres: severe CNS defects, MR, death. Urine smells like maple syrup. |
I Love Vermont MAPLE SYRUP from BRANCHED maple trees.
|
|
|
Deficiency in Hartnup disease??
|
AR
defective neutral AA transporter (tyrosine) on renal and intestinal epithelial cells. CDauses tryp excretion in urne and decreased absorption in gut. Leads to PELLAGRA, cerebellar ataxia, aminoaciduria. Blue diaper syndrome 3 Ds - Dermatitis, Diarrhea, Dementia. |
|
|
|
What are the sx of Pellagra?
|
3Ds -
Diarrhea Dementia Dermatitis (Death) |
|
|
|
Which substrates are required to synthesize purines?
|
Glycine, Aspartate, Glutamine, THF, and CO2
|
GAG + COTH
|
|
|
Which substrates are required for pyramidine synthesis?
|
Glutamine, Aspartate, THF, CO2
|
GA + Coth
|
|
|
Which is the RLS of purine synthesis?
|
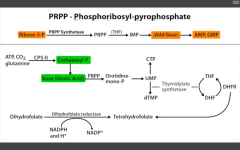
Glutamine PRPP amidotransferase (between PRPP and IMP).
Inhibited by 6-Mercaptopurine. |
|
|
|
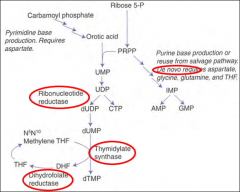
Which step of de novo purine or pyramidine synthesis is inhibited by hydroxyurea?
|
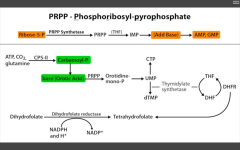
De novo pyramidine synthesis:
Inhibits ribonucleotide eductase, which converts UDP to dUDP. |
|
|
|
Which step of de novo purine or pyramidine synthesis does 5-FU inhibit?
|
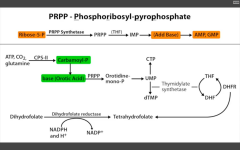
De novo pyramidine synthesis.
Inhibits Thymidylate synthase (converts dUMP --> dTMP) Requires THF, which cycles using Dihydrofolate reductase to be regenerated. |
|
|
|
Which step of de novo purine or pyramidine synthesis does MTX inhibit? For whom?
|
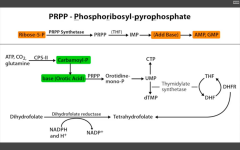
De novo pyramidine synthesis.
DHFR (converts DHF --> THF to be reused as a cofactor for thymidylate synthase (converts dUMP --> dTMP). For eukaryotes. Used as anti-cancer or immunosuppressant in rheumatic disease. |
|
|
|
Which step of de novo purine or pyramidine synthesis does 6-MP inhibit?
|
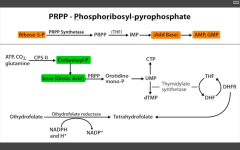
Purine synthesis.
Inhibits RLS - glutamien PRPP amidotransferase. prevents de novo purine synthesis. |
|
|
|
What happens in an Ornithine Transcarbamoylase deficiency?
|
Key enzyme in the urea cycle.
Leads to accumulation of carbamoyl phosphate, which is then converted to orotic acid. With a urea cycle enz deficiency, you have hyperammonemia AND orotic aciduria. |
|
|
|
What happens in orotic aciduria?
|
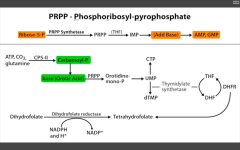
Defect in UMP synthase of de novo pyramidine synthesis pathway. Cannot convert orotic acid to UMP.
Get increased orotic acid in urine, NO hyperammonemia (vs. OTC deficiency), megaloblastic anemia that DN improve with B12 or folate administration (because problem isn't with THF), failure to thrive. Rx: oral uridine administration. |
|
|
|
What is the MOA of etoposide and fluoroquinolones?
|
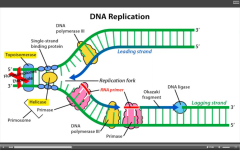
Inhibit eukaryotic and prokaryotic topoisomerase, respectively.
|
|
|
|
What is Anti-scl70 targeting, what dz is it associated with?
|
Targets topoisomerase.
A/w diffuse scleroderma. |
|
|
|
What is defective in xeroderma pigmentosum? What are the clinical manifestations?
|
Defective nucleotide excision repair.
Prevents repair of thymidine dimers d/t UV light exposure. Dry skin, melanoma, hypersensitivity to UV light, moreso than albinism. 1,000 fold increased risk of skin cancer. |
|
|
|
What is defective in HNPCC (hereditary nonpolyposis colorectal cancer)? What are the clinical manifestations?
|
Mutated mismatch repair. increased risk of colon cancer.
|
|
|
|
What is defective in Ataxia telangiectasia?
|
Mutated non-homologous end joining in DNA synthesis/repair.
Sensitivity to ionizing radiation. Cerebellar ataxia, onset 1-2y/o. Telangiectasia, onset 5y/o. Poor smooth pursuit. immunodeficiency associated. Increased alpha-FP in kids >8months old. |
|
|
|
What is defective in Bloom syndrome? What are the clinical manifestations?
|
DNA helicase defect.
Hypersensitivity to light, leukemias, lymphomas, cancer onset at age 25, average. AR, askenazi jews. Biutterfly scaly rash (nose/cheeks), telangiectasias, cafe au lait spots, immunodeficiency. |
|
|
|
What is deficient in BRCA1/BRCA2 mutation? What are the clinical manifestations?
|
Defective repair of DS DNA breaks.
Increased risk of breast/ovarian cancer, specifically. |
|