Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
249 Cards in this Set
- Front
- Back
|
nomenclature of benzene derivatives follows same rules used for
|
other substituted hydrocarbons
|
|
|
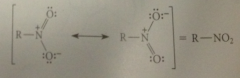
nitro group can be represented as a resonance hybrid of two equiv dipolar structures
|
|
|
|
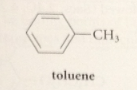
toluene
|
|
|
|
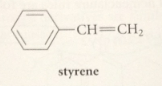
styrene
|
|
|
|
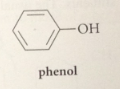
phenol
|
|
|
|
anisole
|
|
|
|
when none of the substituents qualifies as a principal group
|
the substituents are cited & numbered in alphabetical order
|
|
|
If a substituent is eligible for citation as a principal group
|
it is assumed to be at carbon-1 of the ring
|
|
|
o-xylene
|
|
|
|
m-cresol
|
|
|
|
catechol
|
|
|
|
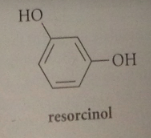
resorcinol
|
|
|
|
hydroquinone
|
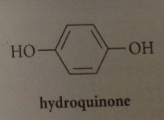
|
|
|
when a benzene derivative contains 2+ substituents on the ring
|
only #s may be used to designate the positions of substituents - usual nomenclature rules followed
|
|
|
is it ever simpler to name a benzene ring as a substituent group?
|
yes
|
|
|
benzene ring or substituted benzene ring cited as sub referred to generally as
|
aryl group
|
|
|
unsub benzene ring as a substituent
|
phenyl group
|
|
|
Ph-Ch2 group
|
benzyl group
|
|
|
bp of benzene derivatives
|
similar to those of other hydrocarbons w similar shapes & mm
|
|
|
mp of benzene & cyclohexane
|
unusually high bc of symmetry
|
|
|
Addition of a C atom adds
|
20-30 C to the bp
|
|
|
mp of p-disub benzene derivatives
|
typically much higher than those of corresponding o/m isomers
|
|
|
bc isomer w highest mp is usually one that most easily crystallized
|
many p-sub cmpds can be separated from o/m isomers by recrystallization
|
|
|
benzene / aromatic density
|
not as dense as water, more dense than alkanes, alkenes of about same mm
|
|
|
benzene & hydrocarbon deriv insoluble in
|
h2o
|
|
|
benzene derivatives w sub that form H-bonds to water
|
more soluble
|
|
|
most useful absorptions in IR spectra of benzene derivatives
|
C-C stretch absorptions of the ring, lower freq than C=C absorption of alkenes
|
|
|
C-C stretch occurs @ lower freq than alkene C=C bc
|
C-C bonds in benzene rings have bond order of 1.5
|
|
|
overtone & combination bands
|
1660-2000 cm-1 (help determine substitution patterns)
|
|
|
proton NMR spectrum of benzene
|
singlet @ chem shift of 7.4
|
|
|
chem shifts are greater than those of alkenes by
|
1.5-2 ppm
|
|
|
NMR absorptions @ large chem shifts
|
particularly characteristic of most benzene derivatives
|
|
|
pi-e density in benzene lies
|
in 2 doughnut-shaped regions above & below plane of the ring
|
|
|
In an NMR experiment
|
benzene mlcs in soln are moving about randomly- assume all possible orientations rel to applied field Bo, but particular orientation dominates chem shift
|
|
|
ring current
|
circulation of pi e around the ring
|
|
|
Ring current induces
|
magnetic field Bi, forms closed loops thru the ring
|
|
|
Induced field opposes applied field
|
along axis of ring, but augments applied field outside ring (region occupied by benzene protons)
|
|
|
Augment field outside ring
|
correspondingly higher freq required for absorption, chem shifts of aromatic protons inc
|
|
|
ring current & large chem shift characteristic of cmpds that are
|
aromatic by Huckel 4n+2 rule
|
|
|
Basis of both ring current & aromaticity
|
overlap of p orbitals in cont cyclic array
|
|
|
when protons in sub benzene derivative nonequiv
|
split each other
|
|
|
can splitting occur across 1+ C-C bond?
|
yes
|
|
|
leaning
|
chem shifts of two coupled protons similar, intensities differ from 1:1 ideal
|
|
|
Two leaning doublet pattern typical of
|
disub benzene rings in which 2 diff ring sub have a para relationship
|
|
|
protons ortho to the more electropositive group
|
have a smaller chem shift
|
|
|
benzylic protons
|
protons on C adjacent to benzene ring
|
|
|
chem shift of benzylic protons
|
2-3 ppm
|
|
|
OH absorptions of phenols
|
lower field than those of alcohols & undergo exchange in D2O
|
|
|
In C NMR spectra, chem shifts of aromatic C
|
in C-C db region 110-160 ppm
|
|
|
Exact value depend on
|
ring substituents present
|
|
|
Chem shift of benzene
|
128.5 ppm
|
|
|
Quaternary ring C
|
higher chem shift (bears no H)
|
|
|
Bc proton-decoupling technique enhances size of peaks of C that bear H
|
peaks for C that do not bear H considerably smaller
|
|
|
chem shifts of benzylic C
|
18-30 region
|
|
|
simple aromatic hydrocarbons: 2 absorption bands in UV spectra
|
rel strong @ 210 nm, weaker near 260 nm
|
|
|
Substituent groups on the ring alter
|
both lambda max values & intensities of both peaks, esp if sub has an unshared e pair or 2p orbitals that can overlap w pi -e system of aromatic ring
|
|
|
Most extensive conj associated w inc in
|
both lambda max & intensity
|
|
|
VSEPR rules predict that O of p-ethylanisole like O of water should be tetrahedral & sp3, but O is sp2
|
allows one e pair to occupy a 2p orbital, which has same size, shape & orientation as the C 2p orbitals of the ring
|
|
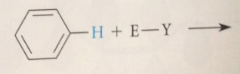
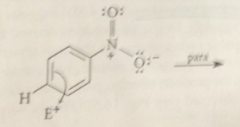
EAS: H of aromatic ring sub by
|
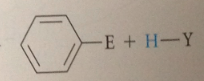
electrophile (lewis acid)
|
|
|
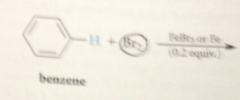
all EAS rxns occur by..
|
similar mech
|
|
|
|
|

First step in mech of benzene bromination
|
formation of complex btwn Br2 & FeBr3
|
|
|
Formation of complex results in
|
formal pos charge on one of bromines
|
|
|
Pos charged bromine
|
better e acceptor, better LG than bromine in Br2 itself
|
|
|
-FeBr4 is a __ base than Br-
|
weaker
|
|
|
-FeBr4 is essentially the prod of
|
a Lewis acid-base association rxn of Br- w FeBr3
|
|
|
In FeBr4 an e pair on Br- has already been donated to
|
Fe, is less available to act as a base than a naked e pair on Br- itself
|
|
|
This step results in formation of resonance stabilized carbocation but
|
disrupts aromatic stabilization of the benzene ring
|
|
|
Harsh conditions (high reagent conditions, high temp, & strong Lewis acid cat) required bc
|
2nd step does not occur under milder conditions used to bring about bromine addition to alkene
|
|
|
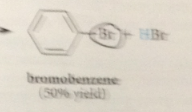
rxn completed when
|
bromide ion (complexed to FeBr3) acts as base to remove ring proton, regenerate cat. FeBr3 & give products bromobenzene & HBr
|
|
|
Rxn of bromide @ e-deficient C itself doesn't occur
|
bc the resulting addition product would not be aromatic - by losing a B-proton instead the carbocation can form bromobenzene a stable aromatic cmpd
|
|
|
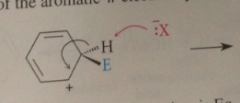
EAS steps
|
generation of an electrophilic, nuc rxn of pi e of the aromatic ring w electrophile to form a resonance-stabilized carbocation intermediate
|
|
|
the electrophile approaches the pi e cloud fo the ring
|
above or below the plane of the mlc
|
|
|
In the carbocation intermediate
|
the C @ which the electrophile reacts becomes sp3 hybridized & tetrahedral
|
|
loss of a proton from the carbocation intermediate to form
|
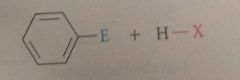
the substituted aromatic cmpd
|
|
|
The proton is lost from the C @ which
|
substitution occurs
|
|
|
This C again becomes
|
part of the aromatic pi e system
|
|

|
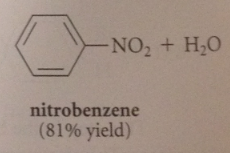
|
|
|
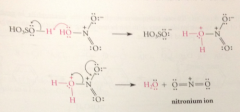
electrophile in nitration
|
+NO2 (the nitronium ion) formed by acid-cat removal of the elements of water from HNO3
|
|
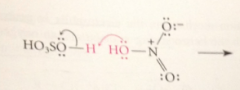
|
|
|
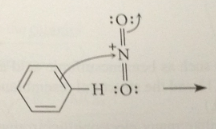
rxn of the benzene pi e w the electrophile
|
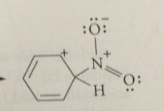
to form a carbocation intermediate
|
|
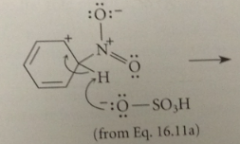
Loss of a proton from the carbocation
|
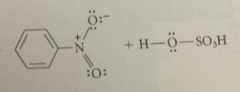
to give a new aromatic cmpd
|
|
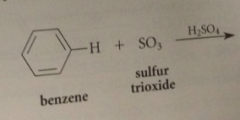
sulfonation
|
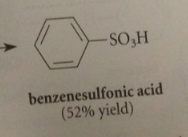
|
|
|
Sulfur trioxide
|
fuming liquid that reacts violently w water to give H2SO4
|
|
|
Source of SO3 for sulfonation
|
usually a soln of SO3 in conc H2SO4 called fuming sulfuric acid or oleum
|
|
|
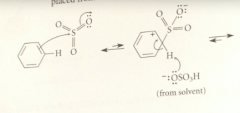
in one sulfonation mech, the electrophile is
|
neutral sulfur trioxide
|
|
|
When sulfur trioxide reacts w the benzene ring pi e
|
an oxygen accepts the e pair displaced from sulfur
|
|
|
|
|
|
Sulfonic acids such as benzenesulfonic acid
|
are rather strong acid
|
|
|
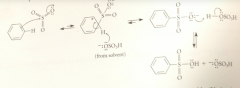
Is sulfonation reversible?
|
Yes
|
|
|
The SO3H group is replaced by H when
|
sulfonic acids are heated w steam
|
|
|
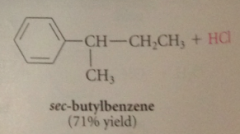
|
|
|
alkylation
|
rxn that results in the transfer of an alkyl group
|
|
|
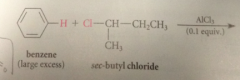
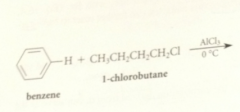
Friedel-Crafts alkylation
|
an alkyl group is transferred to an aromatic ring in the presence of an acid cat
|
|
|
electrophile in FC alkylation formed by
|
complexation of the Lewis acid AlCl3 w the halogen of an alkyl halide
|
|
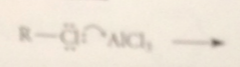
|
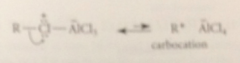
|
|
|
If the alkyl halide is secondary or tertiary
|
complex can further react to form carbocation intermediate
|
|
|
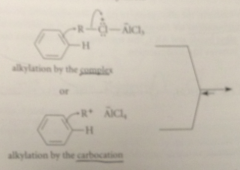
The electrophile in FCA
|
either alkyl halide Lewis acid complex or carbocation derived from it
|
|
|
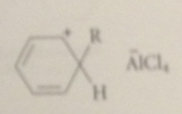
|
|
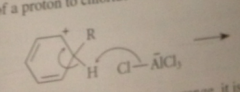
loss of a proton to chloride ion
|
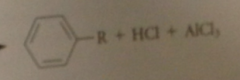
completes the alkylation
|
|
|
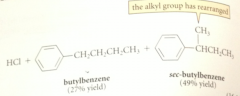
|
|
|
bc primary carbocations are too unstable to be involved as intermediates
|
it is prob the complex of the alkyl halide & AlCl3 that rearrranges - has enough carbocation character that it behaves like a carbocation
|
|
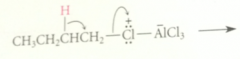
|
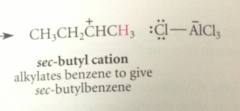
|
|

|
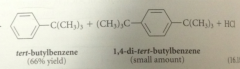
|
|
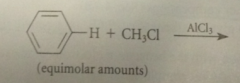
|

|
|
|
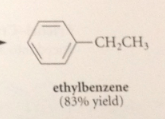
a monoalkylation prod can be obtained in good yield if
|
a large excess of the aromatic sm is used
|
|
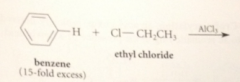
|
|
|
|
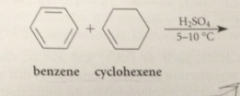
if alkenes and alcohols are used as the alkylating agents in FCA, the carbocation electrophiles are generated
|
from alkenes by protonation & from alcohols by dehydration
|
|
|
|
|
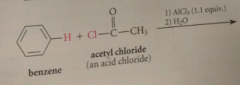
when benzene reacts w an acid chloride in the presence of a lewis acid such as AlCl3
|
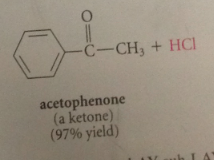
a ketone is formed
|
|
|
acylation rxn
|
acyl group transferred from one group to another
|
|
|
FCA
|
an acyl group is introduced into an aromatic ring in presence of a Lewis acid
|
|
|
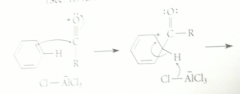
electrophile in FCA
|
carbocation called acylium ion, formed when acid chloride reacts w Lewis acid AlCl3
|
|
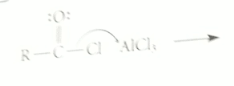
|

|
|
|
Weaker lewis acids such as FeCL3 & ZnCl2 can be used to form acylium ions in FCA
|
of aromatic cmpds that are more reactive than benzene
|
|
|
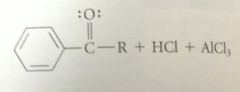
|
|
|
ketones are weakly
|
basic
|
|
|
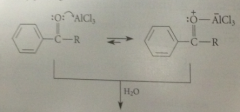
ketone prod of FCAc reacts w Lewis Acid
|
in Lewis acid-base association to form a complex that is catalytically inactive
|
|
|
Consequences of formation of this complex
|
at least one equiv of lewis acid must be used to ensure its presence throughout the rxn, & complex must be destroyed before ketone prod can be isolated
|
|
|
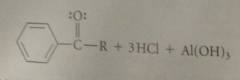
|
|
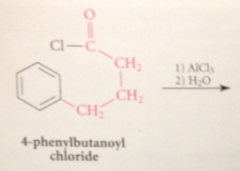
|
|
|
|
Type of rxn can only occur at
|
an adjacent ortho position bc rxn @ other positions would produce highly strained prod
|
|
|
When 5 or 6 membered rings are involved
|
this process is much faster than rxn of acylium ion w the phenyl ring of another mlc
|
|
|
proximity effect
|
kinetic advantage of intramlclr rxns
|
|
|
The multiply sub prod observed in FCA are not a problem in FCacy
|
bc the ketone prod of acylation are much less reactive than the benzene sm
|
|
|
Alkylation rxn is useful for preparing
|
certain alkylbenzenes
|
|
|
ACylation rxn is excellent method for
|
synth of aromatic ketones
|
|
|
When a monosub benzene undergoes an EAS rxn
|
3 possible disub prod might be obtained
|
|
|
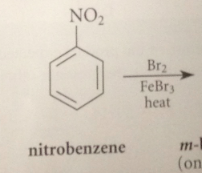
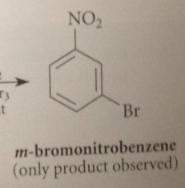
Nitration of bromobenzene could give
|
ortho, meta or para bromonitrobenzene
|
|
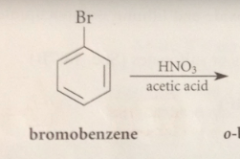
|
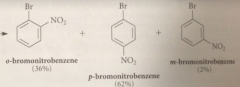
|
|
|
If a sub benzene undergoes further sub mostly @ the ortho and para positions
|
the original sub is called an O, p directing group
|
|
|
Bromine is an o p directing group bc
|
all electrophilic sub rxns of bromobenzene occur @ the o and p positions
|
|
|
|
|
|
Other electrophilic sub rxns of nitrobenzene also give
|
mostly the meta isomers
|
|
|
nitro group is a
|
meta directing group
|
|
|
EAS rxns at 1 position of a benzene derivative
|
are much faster than the same rxns @ another position
|
|
|
Substitution rxns at the diff ring positions are
|
in competition
|
|
|
all o p directing substituents are either
|
alkyl groups or groups that have unshared e pairs on atoms directly attached to the benzene ring
|
|
|
atom directly attached to the benzene ring has
|
unshared e pairs
|
|
|
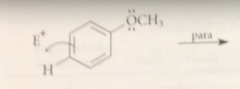
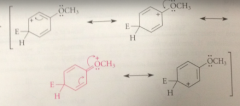
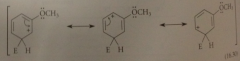
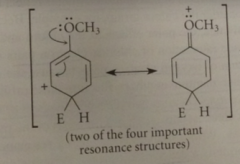
Reaction of E+ at the para position of anisole gives a
|
carbocation intermediate w the following four important resonance structures
|
|
|
|
|
|
the unshared e pair of the methoxy group can
|
delocalize the pos charge on the carbocation
|
|
|
Espec important structure bc it contains
|
more bonds than others & every atom has an octet
|
|
|
if the electrophile reacts w anisole @ the meta position
|
the carbocation intermediate that is formed has fewer resonance structures than the ion
|
|
|
The charge cannot be delocalized onto the
|
OCH3 group when rxn occurs @ the meta position
|
|
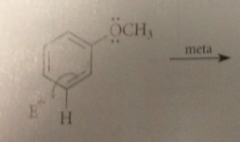
|
|
|
|
For the O to delocalize the charge
|
it must be adjacent to an e deficient C
|
|
|
Pos charge is shared on
|
alternate C of the ring
|
|
|
When meta sub occurs
|
the pos charge is not shared by the C adjacent to the O
|
|
|
Rxn of an electrophile @ either o or p positions of anisole
|
gives a carbocation w more resonance structures (more stable carbocation)
|
|
|
RLS in many EAS rxns is
|
formation of the carbocation intermediate
|
|
|
Prod derived from the more rapidly formed carbocation
|
the more stable carbocation are the ones observed
|
|
|
Substituents containing atoms w unshared e pairs adjacent to the benzene ring are
|
o p directors in EAS rxns bc their e pairs can be involved in the resonance stabilization of the carbocation intermediates
|
|
|
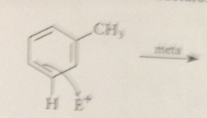
Rxn of E+ @ position o or para to an alkyl group gives
|
an ion that has one tertiary carbocation resonance structure
|
|
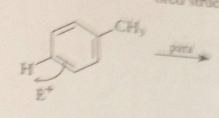
|
|
|
|
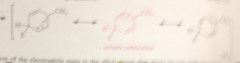
Rxn of the electrophile meta to the alkyl group gives
|
an ion w all resonance forms w secondary carbocations
|
|
|
|
|
|
bc rxn @ the o or p position gives
|
the more stable carbocation, alkyl groups are o p groups
|
|
|
m directing groups are all
|
polar groups that do not have an unshared e pair on an atom adjacent to the benzene ring
|
|
|
|
|
|
|
|
|
Bc repulsion btwn 2 like charges & so E of interaction inc w dec separation
|
the resonance structure is less important than the others
|
|
|
By Hammond's postulate the more stable carbocation intermediate should be
|
formed more rapidly
|
|
|
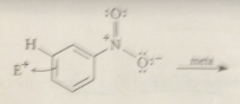
Nitro group is a meta director bc
|
the ion that results from meta sub is more stable than the one that results from para sub
|
|
|
Substituents that have pos charges adjacent to the aromatic ring are
|
meta directors bc meta substitution gives the carbocation intermediate in which like charges are further apart
|
|
|
Not all meta-directing groups have full pos charges like the nitro group but
|
all of them have bond dipoles that place a substantial amt of pos charge next to the benzene ring
|
|
|
aromatic substitution rxn of a benzene derivative bearing an o, p directing group would give
|
2 x o as p prod if substitution were completely random bc there are 2 o positions
|
|
|
which sub prod is major one in rxn mixture?
|
para
|
|
|
FCac of toluene gives essentially
|
all para substitution prod & almost no ortho prod
|
|
|
The electrophile cannot react @ the o position w/o developing
|
VDW repulsions w the methyl group that is already on the ring
|
|
|
nitration of toluene gives
|
twice as much o-nitrotoluene as p-nitrotoluene
|
|
|
Nitration of toluene @ either o or p position is
|
so fast that it occurs on every encounter of the reagents
|
|
|
Ready availability of o-nitrotoluene makes it
|
a good sm for certain o-sub benzene derivatives
|
|
|
If EAS rxn yields a mix of o and p isomers
|
a problem of isomer separation arises that mus tbe solved if the rxn is to be useful
|
|
|
para isomer of o, p pair typically has
|
the higher melting point
|
|
|
Activating group
|
a substituted benzene derivative reacts more rapidly than benzene itself
|
|
|
deactivating group
|
substituted benzene derivative reacts more slowly than benzene itself
|
|
|
A given substituent group is either
|
activating in all EAS rxns or deactivating in all such rxns
|
|
|
all meta-directing groups are
|
deactivating groups
|
|
|
all o, p directing groups except for halogens
|
are activating groups
|
|
|
Halogens are
|
deactivating groups
|
|
|
Directing effects are concerned w
|
the relative rates of substitution @ diff positions of the same cmpd
|
|
|
Activating or deactivating effects are concerned w
|
the relative rates of substitution of diff cmpds
|
|
|
Consider the effect of the substituent on the
|
stability of the intermediate carbocation, then apply Hammond's postulate by assuming that the stability of this carbocation is related to the stability of the TS for its formation
|
|
|
resonance effect of a sub group
|
ability of the sub to stabilize the carbocation intermediate in electrophilic substitution by delocalization of e from the substituent into the ring
|
|
|
The resonance effect is the same effect responsible for the
|
o, p directing effects of substituents w unshared e pairs i.e. OCH3 & halogen
|
|
|
resonance effect of the methoxy group stabilizes the carbocation
|
|
|
|
polar effect
|
tendency of sub group by virtue of its electronegativity to pull e away from the ring
|
|
|
When a ring substituent is electronegative
|
it pulls e of the ring toward itself & creates e deficiency or pos charge in the ring
|
|
|
In carbocation intermediate of an electrophilic sub rxn
|

pos end of the bond dipole interacts repulsively w the pos charge in the ring, raising the E of the ion
|
|
|
e donating resonance effect of a substituent group w unshared e pairs
|
if it were dominant, would stabilize pos charge & would activate further substitution
|
|
|
If such a group is electroneg, its EWD polar effect, if dominant
|
would destabilize pos charge & would deactivate further substitution
|
|
|
Whether a substituted derivative of benzene is activated or deactivated toward further substitution
|
depends ont he balance of the resonance and polar effects of the substituent group
|
|
|
Anisole undergoes elec sub more rapidly than benzene bc
|
the resonance effect of the methoxy group far outweights its polar effect
|
|
|
The benzene mlc
|
has no sub to help stabilize the carbocation intermediate by resonance
|
|
|
Carbocation intermediate (& TS) derived from the elec sub of anisole is
|
more stable relative to sm than the carbocation (& TS) derived from the elec sub of benzene
|
|
|
In a given rxn, the o and p sub of anisole
|
are faster than the sub of benzene
|
|
|
The methoxy group activates
|
the benzene ring toward o and p substitution
|
|
|
altho the o and p positions of anisole are highly activated toward sub
|
the meta position is deactivated
|
|
|
When sub occurs in the meta position
|
the methoxy group cannot exert its resonance effect & only its rate retarding polar effect is operative
|
|
|
Whether a group activates or deactivates further sub depends on
|
the position on the ring being considered
|
|
|
The methoxy group activates
|
o, p sub & deactivates meta sub
|
|
|
bc o p sub is the observed mode of substitution
|
the methoxy group is considered to be an activating group
|
|
|
the deactivating effects of halogen substituents reflect
|
a diff balance of resonance and polar effects
|
|
|
resonance interaction of chlorine e pairs w the ring
|
is much less effective than the interaction of o e pairs bc the chlorine valence e reside in orbitals w higher quantum numbers
|
|
|
Bc these orbitals & the C 2p orbitals of the benzene ring have diff sizes & diff #s of nodes
|
they do not overlap so effectively
|
|
|
w a weak rate-enhancing resonance effect & strong rate-retarding polar effect
|
chlorine is a deactivating group
|
|
|
Bromine & iodine exert weaker polar effects than chlorine but
|
their resonance effects are also weaker, so they are deactivating groups
|
|
|
Fluorine as a second period element has a stronger resonance effect than the other halogens
|
but as the most electroneg element it has a stronger polar effect as well - deactivating
|
|
|
The deactivating, rate-retarding polar effects of the halogens are similar at all ring positions but offset somewhat by
|
their resonance effects when substitution occurs para to the halogen
|
|
|
Resonance effect of a halogen cannot come into play when
|
substitution occurs @ the meta position of a halobenzene
|
|
|
Meta substitution in halobenzene is deactived
|
even more than para substitution
|
|
|
Alkyl sub such as methyl group have no resonance effect but
|
polar effect of an alkyl group toward e deficient C is an electropos, stabilizing effect
|
|
|
Alkyl sub on a benzene ring stabilize carbocation intermediates in elec sub
|
so they are activating groups
|
|
|
Bc a nitro group has no e donating resonance effect
|
the polar effect of this electroneg group destabilizes the carbocation intermediate & retards elec sub @ all positions of the ring
|
|
|
nitro group is a meta directing group bc
|
sub is retarded more at the o and p positions than at the meta positions
|
|
|
the meta directing effect of the nitro group is not due to selective activation of the meta positions but
|
to greater deactivation of the o and p positions
|
|
|
When an elec sub rxn is carried out on a benzene derivative w more than one substituent
|
the activating & directing effects are roughly the sum of the effects of the separate substituents
|
|
|
|
|
|
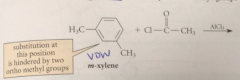
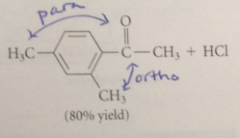
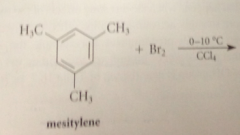
In the FC acylation of m-xylene, both methyl groups
|
direct the substitution to the same positions
|
|
|
methyl groups are
|
o, p directors
|
|
|
Substitution @ the position o to both methyl groups is diff bc
|
VDW repulsions btwn both methyls & the electrophile would be present in the TS
|
|
|
Substitution occurs at a ring position that is
|
para to one methyl & ortho to other
|
|
|
|
|
|
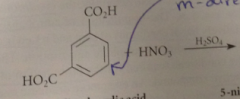
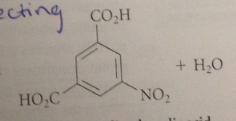
Two meta directing groups on a ring direct further substitution to
|
the remaining open meta position
|
|
|
if one group is much more strongly activating than the other
|
the directing effect of the more powerful activating group generally predominates
|
|
|
|
|
|
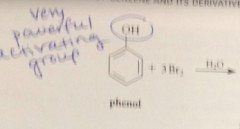
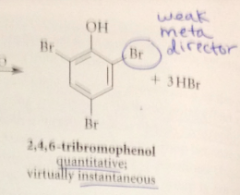
after the first bromination, the OH & Br groups direct subsequent brominations
|
to diff positions
|
|
|
strong activating & directing effect of the OH group @ ortho & para
|
overrides the weaker directing effect of the Br group
|
|
|
|
|
|
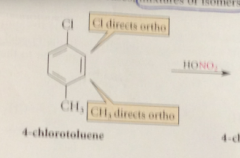
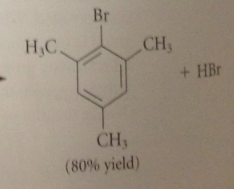
activating or deactivating effects of substituents in an aromatic cmpd determine
|
the conditions that must be used in an elec sub rxn
|
|
|
|
|
|
|
|
|
when a deactivating group is being introduced by an elec sub rxn
|
it is easy to introduce one gorup @ a time bc the products are less reactive than the reactants
|
|
|
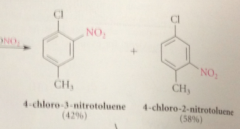
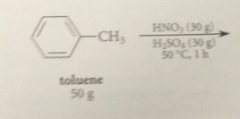
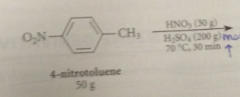
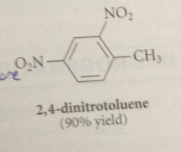
toluene can be nitrated only once bc
|
the nitro group that is introduced retards a second nitration on the same ring
|
|
|

|
|
|
|
|
|
|
|
|
when an activating group is introduced by elec sub
|
the prod are more reactive than the reactants
|
|
|
additional sub can occur easily under the conditions of the first substitution
|
so mixtures of prod are obtained
|
|
|
some deactivating substituents
|
retard some rxns to the point they are not useful
|
|
|
|
|
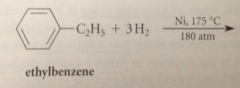
|
|
|
|
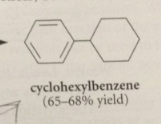
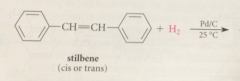
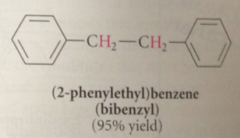
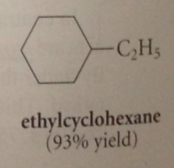
a good way to prepare a substituted cyclohexane
|
prepare corresponding benzene derivative, then hydrogenate it
|
|
|
cat. hydrogenation of benzene derivatives gives
|
corresponding cyclohexanes & cannot be stopped at the cyclohexadiene or cyclohexane stage
|
|
|
The delta H of hydrogenation of benzene can be used to
|
provide another estimate of aromatic stabilization E of benzene
|
|
|
bc the first hydrogenation rxn of benzene is endothermic
|
E must be added for it to take place
|
|

|
|
|
|
|