Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
46 Cards in this Set
- Front
- Back
|
Why understand cancer genetics?
|
Diagnostics, prognostics, stratification, therapeutic targeting
|
|
|
Strategy to prevent recurrent cancer
|
Targeted cancer treatment based on individual gene expression patterns
|
|
|
Malignant cell transformation
|
State change from normal cell to a cancer cell; Malignant cancer cells invade neighboring tissues, enter blood vessels and metastasize to different sites; caused by several mutations, not just one.
|
|
|
Benign hyperplastic cells
|
Tumor cells grow only locally and cannot spread by invasion or metastasis
|
|
|
Normal cell division vs. Cancer cell division
|
Normal: cell damage without repair--> apoptosis; Cancer: mutation--> passes on to daughter cells, 2nd mutation--> passes on to daughter cells, 3rd mutation---> uncontrolled growth
|
|
|
Common features of cancer cells (12)
|
(1) Sustained angiogenesis (2) tissue invasion & metastasis (3) Evading apoptosis (4) Self-sufficiency in growth signal (5) insensitivity to anti-growth signals (6) limitless replicative potential (7) DNA damage stress (8) Oxidative stress (9) Mitotic stress (10) Proteotoxic stress (11) Metabolic stress (12) Evading immune surveillance....basically cancer cells live a stressful life
|
|
|
Causes of cancer (8)
|
(1) Viruses & bacteria- example: Hepatitis B, cervical cancer (Gardasil vaccine) (2) Chemical exposure (3) Radiation exposure (4) Hereditary (5) Diet (6) Hormones (7) Oncogene & Tumor suppressor mutation (8) Random accumulation of mutations due to DNA replication & oxidative stress--this is why we can't prevent all cases because we can't stop mutations from happening
|
|
|
Somatic mutations
|
Occur in nongermline tissues; are nonheritable
|
|
|
Germline mutations
|
Present in egg or sperm; are heritable- all cells affected in offspring; cause cancer family syndrome
|
|
|
Cancer cells have mutations in:
|
primarily: oncogenes, tumor suppressor genes, DNA repair genes;
also: cell death genes, cell signaling genes, cell checkpoint genes, cell senescence genes, cell differentiation genes, metastasis/invasion genes |
|
|
Oncogene
|
gene mutated in cancer, whose increased expression or activity drives cell transformation; these get turned on in cancer
|
|
|
Tumor suppressor
|
gene mutated in cancer, whose decreased expression or activity allows cell transformation; are inactive in cancer
|
|
|
Stages where mutations can occur
|
growth factors, receptors (Her2, EGFR), signaling enzymes (Ras, Raf), transcription factors (Myc, NF-kB)
|
|
|
Gene amplification
|
leads to overexpression of gene which leads to more signaling than you should actually be getting, leads to insensitivity
|
|
|
Translocation of genes
|
example: in leukemia, Bcr-Abl genes, leads to higher expression of abl
|
|
|
Genes that act like brake pedals
|
Tumor suppresor genes; can inhibit at each stage: receptor--->signaling enzymes-->transcription factors; also if the recycling of receptors is inhibited, this inhibits receptor
|
|
|
Tumorigenesis
|
Caused by the loss of BOTH copies of tumor suppressor (called loss of heterozygosity event)
|
|
|
p53
|
A tumor suppressor gene that triggers cell suicide and regulates apoptosis; usually mutated in cancer cells which causes inappropriate cell survival; looking for a way to change mutant back to non-mutant
|
|
|
Oncogene Cooperation
|
Interaction between oncogene mutations causes the emergence of malignant transformation; cancer doesn't happen with just one mutation of oncogene or of one tumor suppressor; 3-5 mutations cause cancer; the cooperation drives malignant transformation; Just Ras mutation or just Myc mutation doesn't cause cancer, but mutation in both causes cancer
|
|
|
Cancer genome landscapes
|
Show how many mutations are in each gene; certain genes have many mutations (p53)
|
|
|
Predicted rates of colon cancer based on 1, 2 or 4 mutations
|
If only one mutation caused cancer, there would be a linear relationship between age and risk of cancer. The more mutations needed to cause cancer, the more curved the graph becomes. Comparing to actual data shows that 3-5 mutations cause cancer
|
|
|
Cancer cells have complex architecture
|
They rely on changes in effectors which underlies malignant transformation:
A +B -----> invasion, growth, angiogenesis, survival ---> transformation; NOT: A ----> Invasion, angiogenesis ---> transformation; B -----> survival, growth -----> transformation; |
|
|
Two functions of Myc; Cooperation of Myc with Bcl-2
|
(1) Myc-Max ----> Proliferation
(2) Myc-Max -----> Death; inhibited by Bcl-2 & IGF1; In normal cells, Myc causes cell death |
|
|
Cooperation of mutant Ras/Raf with p53
|
-Promotes cell proliferation
-Ras/Raf + p53 ----> p21 -----| cyclin cdk; (p21 inhibits growth arrest) -Ras/Raf alone ------------> cyclin cdk on |
|
|
Cooperation response genes
|
Essential mediators of malignant transformation synergistically (more than additive) regulated in response to the combination of loss-of-function mutant p53 and constitutively active Ras. Changes in apoptosis, transcription, signaling, metabolism, & adhesion.
|
|
|
What do DNA tumor viruses do?
|
They alter cellular gene function; example: HPV, Epstein-Barr, chronic infection
|
|
|
What do transforming retroviruses do? What did Bishop & Varmus find?
|
They acquire and transmit cellular DNA.
B & V showed that gene sequences in transforming retroviruses were homologous to sequences in cellular DNA; Example: Rous Sarcoma virus (Src); myelocytomatosis virus (myc); |
|
|
p53: tumor suppressor or oncogene?
|
Oncogene evidence: (1) adding anti p53 to mouse 3T3 cells causes growth arrest (2) in rat embryonic fibroblasts, p53 cDNA immortilizes cell and p53 cDNA + Ha-Ras causes transformation into tumor. Experiment lacks loss of function control.
Tumor suppressor evidence: Mutant (& inactive) p53 found in tumor cells, the 2nd allele is either lost through loss of the chromosome, or inactived by deletion. This seems to confer a selective growth advantage to cells Answer: wild type p53 is a tumor suppressor, mutant is an oncogene. Why was there oncogene evidence? the cDNA was for a mutated p53 |
|
|
p53 and cancer
|
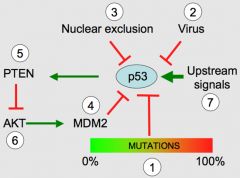
1- p53 mutations can be found in 50% of human cancers, but their penetrance is highly heterogeneous, as reflected by the diverse remaining transactivation activity that ranges from O to 100%. 2- Various DNA viruses, such as SV40, HPV or adenoviruses, encode proteins that target p53 protein. 3- In inflammatory breast cancer and neuroblastoma, p53 is predominantly found in the cytoplasm. 4- mdm2 accumulation is found in numerous cancers, such as sarcoma or breast carcinomas. 5- PTEN, a p53 regulated gene, is mutated in various types of cancer including glioblastoma. 6- Although no mutation of AKT has been found in human cancer, constitutive activation of its kinase activity has been observed via deregulation of the upstream pathway. 7- Mutations in various pathways upstream of p53 (ATM, p19ARF or Hcdk2 gene) can be observed in various types of cancer
|
|
|
What does Rb do?
|
Inhibits cell cycle
|
|
|
Telomerase
|
The reactivation of telomerase is the key to unlock human cell transformation--immortality, because it maintains chromosome length. Without it, telomeres are shortened after each cell division so after a certain number of divisions, the cell dies.
|
|
|
Gleevec
|
A molecular targeted therapy that inhibits BCR-Abl. It's highly selective for tumors with this translocation and has a strong selective pressure for insensitivity. Gleevec binds to BCR-Abl ATP binding site preventing it from phosphorylating
|
|
|
Herceptin
|
A molecular targeted therapy. It's an antibody antagonizing Her2 receptor activation in breast cancer; inhibits growth of Her2+ tumors
|
|
|
Iressa
|
A molecular targeted therapy. It's an EGFR antagonist, turns out it only works against very specific mutant EGFR forms
|
|
|
Extrinsic factors that can be essential for cancer growth (2)
|
(1) Blood supply (angiogenesis & neovascularization)
(2) Hormone signaling (androgen or estrogen dependence) |
|
|
Angiogenesis
|
Tumors secrete factors that drive formation of new blood vessels. Blocking developement of new blood vessels inhibits tumor development and spreading.
|
|
|
Tomoxifen
|
Drug that targets estrogen hormone dependence by blocking ligand binding
|
|
|
Flutamide
|
Drug that targets testosterone hormone dependence of prostate cancer by blocking cofactor interaction
|
|
|
Metastasis
|
Dissemination or spread of cancer cells to distance locations within the body; a hallmark of malignant disease
|
|
|
Seed & Soil hypothesis
|
Hypothesis that some organs are congenial for tumor cells; cancer cells metastasize in target organs due to certain genetic factors
|
|
|
Dominant circulation hypothesis
|
Hypothesis that cancer spreads based on circulation; cancer cells target first highly vascularized organ encountered. Lungs are a major target- one of the first highly vascularized organ encountered, but why not the heart?
|
|
|
Evolution models of where cancer cells spread
|
Selection of rare clones in specific organs by random acquisition of pro-metastatic mutations
|
|
|
Pre-determination models of where cancer cells spread
|
Poor-prognosis genes expressed in primary tumors support primary tumor and distant metastasis
|
|
|
Measuring acquisition of Metastatic capability with gene expression programs
|
Use microarrays to measure differences in gene expression. Upregulated combinations of genes that were upregulated in metastasis and saw this caused more metastasis.
|
|
|
Cancer stem cell/tumor initiating cell
|
Cells within tumor population that are transplantable and produce heterogeneous progeny. They are the drivers of metastasis and tumor recurrence.
|
|
|
What type of disease is cancer?
|
A cellular disorder. There are transforming viruses that carry oncogenes which are cellular in origin.
|