Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
219 Cards in this Set
- Front
- Back
|
Mylanta
|
Class: Antacid
- combination of Al(OH)3 and Mg(OH)2 - generally not absorbed, act locally - adverse effects: hypercalcemia with calcium salts (calcium carbonate: Tums), hypophosphatemia (complex of aluminum with phosphate), impaired acid-dependent absorption of food and drugs |
|
|
Sucralfate
|
class: mucosal protectant
mechanism: binds membranes of epithelial cells forming artificial mucous, protects cells that have damaged/loss mucous membrane creating new barrier - chemistry: modified sucrose to have sulfates and aluminum salts - action: protects mucosa from damage by attack by acid and pepsin: binds proteins of cell surface, selectively binds to damaged tissue, binds pepsin and bile salts (which are irritants), indirectly suppresses growth of H. pylori (interferes with binding to cell surface) - few adverse effects (not absorbed. eliminated by the fecal route) |
|
|
peptobismol
|
class: colloidal bisumuth compound, mucosal protectant
- barrier protection |
|
|
Misoprotsol (Cytotect)
|
class: prostaglandin, mucosal protectant
- mechanism: activate prostaglandin receptors on parietal cells (and other cells) stimulating production of mucosa, which are protective |
|
|
Cimetidine
|
Class: selective histamine receptor antagonist
- blocks acid secretion from parietal cells - inhibits a cytochrome P450: lots of drug and food interactions, needs to be modulated in patients with liver disease - antiandrogenic effect: causing gynecomastia |
|
|
Ranitidine, Famotidine, Nizatidine
|
Class: H2 receptor antagonist (2nd generation)
- block acid secretion from parietal cells - lack antiandrogenic effects and do not interfere with Cytochrome P450 metabolism |
|
|
Omeprazole
|
Class: Proton pump inhibitor
Mechanism: block H+/K+ ATPase (covalently binds and inhibits the activity of the enzyme) - has sustained duration of action (covalent bonding) - prodrug, only active at low pH - decreases H+ secretion - P450 substrate (P450 inhibitors interfere with mechanism of action) - minimal adverse reactions: safe to use |
|
|
Lansoprazole, rabeprazole, esomeprazole
|
Class: proton pump inhibitors
- all similar to omeprazole - differing durations of actions - all pro-drug, activated at low pH - differing side effects and tolerability |
|
|
PPI vs. H2 antagnoists for GERD
|
- Meta-analysis of 134 triaIs involving 35978 esophagitis participants
- Both PPI’s and H2 receptor antagonists are far better than placebo - PPI’s resolve esophagitis and symptoms in higher fraction of patients than do H2 receptor antagonists - Beneficial effects of doubling PPI dose are modest but significant |
|
|
Treatment of H. Pylori
|
Always involves multidrug therapy: PPI + 2 antibiotics
Rationale: PPI allows for healing, 2 antibiotics have complementary action to eliminate infection First line: omeprazole + amoxicillin + clarithromycin Rescue therapy: omeprazole _ bismuth + tetracycline + Flagyl Other antiobiotic: Levofloxacin, rifabutin |
|
|
Germ layer origins of the major components of the gut
|
Endoderm: the epithelial layer of the pharynx, esophagus, stomach, bowel, proximal 2/3 of anus, glandual structures (salivary glands), liver, pancreas, and gall bladder
Ectoderm: oral cavity, distal 1/3 of the anal canal, nerve plexuses Mesoderm: mesothelium, serosa, inner and outer muscle layers, submucosa, muscularis mucosa, lamina propria, lymph |
|
|
Major morphologic events of the gut formation
|
- Around week3 the endoderm folds forming a tube (with yolk stalk hanging. Diverticula begin to bud off to form organs
- cranial and caudal membranes rupture around week 8 forming mouth and anus - Mesoderm then grows around the tube from the splanchnic layer of mesoderm. The somatic layer becomes the parietal layer on the abdominal wall, and the coelom becomes the peritoneum - The primary mesentary suspends the gut from the dorsal wall (dorsal mesentery) and connects it to the ventral wall (ventral mesentery). The ventral mesentery then regresses. Rotation and retroperitoneal positioning then occurs - the stomach rotates bringing the left to the front and the greater curvature caudally - the pancreas and duodenum become positioned retroperitoneally - the gut grows fast into the umbillicus and rotates around the superior mesenteric artery bringing the distal part above the proximal |
|
|
Periodontal disease
|
= disease that attachs the gum and bones around the teeth
- caused by dental plaque, bacteria and host infections - Results in loss of tissues supporting the teeth: gingiva, periodontal ligament (fibrous membrane between teeth), bone, cementum (modified bone around roots) - affects ~65% of the populatin - usually slowly progressive, rapid progression can indicated associated immune deficiency - other diseases have periodontal manifestations: Chediak-Higashi syndrome, chronic granulomatous disease, severe combined immune deficiency, neutropenia, lazy leukocyte syndrome, DM, AIDS |
|
|
Odontogenic infections
|
= infections within and around the teeth, usually secondary to dental caries
- typically due to mixed microflora Mandibular infection sequela: - usually involves the 3rd molars/wisdom teeth. - Infection occurs in lingual aspect of bone (thinnest) and involves the submandibular space - can cause Ludwig's Angina: feeling of choking due to airway compromise from inflammation - causes swelling of the lateral neck and chin Maxillary infections: (less common but more serious) - usually involves anterior teeth (canines) - can spread along the root to the midface and involve facial veins and cavernous sinus: potential cavernous sinus thrombosis - causes midface swelling: upper lip, cheeks, under eye area |
|
|
Systemic manifestations of oral/periodontal infections
|
- Bacterial endocarditis from bacteremia induced by dental procedures: strep viridans (group A) most common, infrequently HACEK bacteria
- CNS lesions from septic emboli (from endocarditis) or CNS abscesses containing oral/nasal flora - infections in other organs are less common |
|
|
Oral HSV infection
|
- antiobodies are present in 90% of the US population
- both HSV-1 and HSV-2 are isolated, dormant virus resides in V sensory ganglion, clinically indistinguishable from varicalla zoster - Only 1-2% of exposed will have primary infection (primary herpetic gingivostomatitis): usually in young children when first exposed, causes painful vessicles (on lips/buccal mucosa, gingiva, palate and tongue), high fever, cervical lymphadenopathy, dehydration risk. Usually resolves ~10-20 days, acyclovir can speed resolution - Secondary painful lesions can occur in up to 40% of the population: typically in along the vermillion border of lips or intraorally exlusively on the attached gingiva, hard palate, or tongue (not movable mucosa). CAn involve the eye if VI nerve is affected. Can be treated with acyclovir prophylactically. Immunocompromised patients have more widespread/variable lesions |
|
|
Aphthous stomatitis
|
= painful oral ulcer in the mouth or upper throat characterized by a break in the mucous membrane with an erythematous ring.
- Affect 20-60% of the US population: higher in upper socioeconomic groups, immune compromised patients have more severe lesions - No identified etiologic agent - Occur on movable mucosa: labial/buccal mucosa, soft palate, floor of mouth - Association with IBD |
|
|
Oral squamous cell carcinoma
|
Epi: most common malignant neoplasm in the upper respiratory tract, but only 2-4% of all malignancies in the US (M>>F due to smoking)
- Common sites: lateral borders and ventral side of the tongue (most common), floor of the mouth, soft palate, gingiva and buccal mucosa (less common) - 5yr survival is 20-60%: stage dependent survival, most patient present late and have poor prognosis Etiology: tobacco (smoking or chewing), HPV infection (better prognosis than others), alcohol, syphilis, Plummer-Vinson syndrome, betel leaf (Asia), viral, genetic Treatment: surgical (large removal of oral tissue), adjuvant radiation/chemo depending on site Types: ulcerative, exophytic, plaque-like Premalignant lesions: leukoplakia (white patch, most are benign, malignant transformation ~20%), erythroplakia (red patch large and persistent, very uncommon almost always associated w/ SCC) |
|
|
Salivary neoplasms
|
Overall: parotid 65-80%, Submandibular 10%, sublingual <5%, other 5-10%
Benign types: - pleomorphic adenoma: most common, mostly parotid (some other small glands), variable histology - Warthin tumor: parotid tumor, most common bilateral tumor (~10%), more common in smokers Malignant types: - mucoepidermoid carcinoma: most common, 60-90% are in parotid or minor glands, much better prognosis if caught early, histologically have large mucin deposits and some epithelial component - Adenoid cystic carcinoma: mostly minor glands, invasive growth pattern around nerves, good short term prognosis but poor long term because of recurrence (nerve involvement) Of glands: parotid mostly benign (70-80%), submandibular 50/50, sublingual mostly malignant (80-90%) |
|
|
Ondontogenic tumors
|
= intraosseous jaw tumors from odontogenic (tooth forming) tissues
- rare, majority are benign, mandibular are most common Amelobastoma: - from enamel forming tissue - has predilection for the posterior mandible - benign but locally destructive (occassionally fatal if maxillary) - treated by en-block resection |
|
|
Paranasal sinus tumors
|
- most commonly occur in the maxillary sinuses but generally rare (both benign and malignant)
Squamous cell carcinoma: most common sinus malignancy, affect maxillary sinus Adenocardinoma: uncommon, can affect nasal cavity and maxillary sinus Predisposing agents for both: smoking, nickel/chromium (dental work), wood working (hard wood: adenocarcinoma, soft wood SCC) |
|
|
Phases of deglutition
|
1. Oral preparatory phase: bolus mastication + mixing w/ saliva, bolus formation
2. Oral voluntary phase: bolus propelled from oral cavity into pharynx -- tongue raised to occlude anterior oral cavity and propel bolus towards oropharynx 3. Pharyngeal phase: bolus transport form oropharynx into esophagus w/out aspiration (requires pharyngeal closing and larynx elevation and closure). Impaired pharyngeal phase → dysphagia, choking, coughing (if airway not protected) 4. Esophageal phase: bolus propelled length of esophagus into stomach; starts w/ the relaxation of cricopharyngeus and opening of UES Contractions: - Primary peristalsis reflex stimulated by swallowing - Secondary peristalsis: stimulated by distension of esophagus (via stretch receptors); occurs if the bolus gets stuck or moves slower than the primary peristaltic wave – secondary waves continue indefinitely until bolus enters stomach - Tertiary peristalsis: not important; maybe related to muscle spasms; may present w/ dysphasia if spasms are frequent |
|
|
Esophageal sphincters
|
- found at either end of the esophagus
- both are tonically contracted to prevent reflux and must recieve inhibitory neural stimulation to relax UES: relaxation allows food to enter the esophagus. LES: physiologic (not a functional structure), relaxes to allow food into the stomach. Stiff LES causes dysphagia, compromised LES causes reflux |
|
|
Techniques to evaluate esophageal disorders
|
- History – “most important”
- Physical Exam: often unremarkable in patients w/ dysphagia; check for signs of systemic abnormalities (dermatologic, endocrine, etc) that prevent w/ dysphagia, e.g. lupus. Check mouth, feel neck/lymph, asses thyroid gland during swallowing- Barium Swallow w/ Xray; asses esophageal anatomy, can determine mechanical dysfunctions (structure, web, diverticulum, etc) -- best initial test for a patient that presents w/ chronic dysphagia (> 3 weeks) - Endoscopy: best to evaluate esophageal muscosa -- initial test w/ acute dysphagia (r/o impaction) - Esophageal manometry: gold standard for suspected motility problems; measures esophageal pressure/contraction/peristalsis |
|
|
Structural disorders of the esophagus
|
- Cervical web: thickened band of the esophageal mucosa causing obstruction
- Zenker’s diverticulum (pharyngoesophageal diverticulus): ballooning of the pharyngeal wall caused by improper opening of the UES - Cricopharyngeal Bars: narrowing due to prominent cricopharyngeus muscle; frequently asymptomatic, myotomy can relieve dysphagia - Cervical osteophytes: bony spurs of the cervical spine (usually elderly patients) push on the esophagus causing an obstruction. Surgery can reduce spurs in severe dysphagia - Esophageal neoplasm: presents with progressive dysphagia, chest pain, cough, weight loss, hematemesis. May be SCC (most common; smoking & alcohol related), Adenocarcinoma (GERD/Barrett’s, inverse relationship w/ H. pylori), Small cell carcinoma, melanoma less common |
|
|
Classic symptoms of esophageal dysfunction
|
heartburn
chest pain regurgitation dysphagia (liquids, solids, both) odynophagia (pain on swallowing) – results from dysfunction of peristalsis or sphincter function; usually presents w/ mucosal abnormalities (irritation, ulceration, etc) |
|
|
Motility disorders of the esophagus
|
Achlasia (aperistalsis): involves smooth muscle layer and LES; characterized by incomplete LES relaxation, increased LES tone, and lack of peristalsis of the esophagus. Presents w/ dysphagia to liquids and solids, regurgitation, chest pain, cough, heartburn. Treated w/ Ca++ blockers, botox, dilation, heller myotomy
- Diffuse esophageal spasm: simultaneous contractions w/in the esophagus resulting in ineffective bolus movement - Nutcracker esophagus: hypertensive/high amplitude contractions, often caused by GERD - Hypertensive lower esophageal sphincter - Ineffective esophageal motility: primary disorder in GERD |
|
|
GERD
|
Pathophys: any mechanism that leads to impaired esophageal mucosal resistance. Incl: defective barrier (low LES pressure/relaxation), reduced esophageal clearance (peristalsis, saliva, position), gastric abnormalities (excess acid, delayed emptying, distention), external factors: diet, smoking, medication)
Symptoms: - Typical: heart burn, regurgitation, dysphagia, water brash (hypersalivation) - Atypical: laryngitis, chronic cough, asthma, dental erosions, laryngeal cancer Complications: esophagitis, esophageal stricture, Barrett's esophagitis (gastric mucosal dysplasia, leading to adenocarcinoma) Diagnosis: PPI trial, endoscopy (only 50% sensitive), ambulatory pH monitoring Therapy:life style modification (avoid triggers, delay laying down after eating, small frequent meals, smoking cessation), medication (H2 blockers, PPI), surgery (Nissen Fundoplication) |
|
|
Esophageal diverticula
|
Upper: Zenker's diverticulum
Middle: Traction diverticulum Lower: Epiphrenic diverticulum These can collect food and grow causing halitosis. Often otherwise asymptomatic |
|
|
Tracheoesophageal fistula
|
= connection between the trachea and the esophagus
- Most commonly congenital, and associated with an atretic segment - Can be caused by surgeries or tumors that erode the walls. - Can result in aspiration of food |
|
|
Mallory-Weiss syndrome
|
= longitudinal tears at the GEJ resulting from severe retching or vomiting.
- Chronic alcoholics, bulimics, or those with acute, severe GI illness are prone to this - Bleeding is not typically severe, but can occasionally be life threatening. - Most patients have co-existing hiatial hernia: herniation of the upper stomach into the thorax through a tear in the diaphragm - Presents with hematemasis |
|
|
Esophageal varices
|
= extremely dilated sub-mucosal veins in the lower 3rd of the esophagus, typically a consequence of portal hypertension
- they are collateral connections between the splanchnic venous circulation and the systemic (normally drains the esophagus into both) - Because they are so superficial they are at high risk for rupture and severe bleeding into the lumen of the esophagus (potentially fatal) |
|
|
Esophagitis
|
= inflammation of the esophagus
- most commonly caused by GERD or infectious agents (candida in immune suppressed, HSV, CMV). Also caused by mucosal irritants (alcohol, acids, alkalis, hot fluids) |
|
|
SCC of the esophagus
|
Epi: 90% of global esophageal cancer. 6/100K in US, 100/100K in Asia
Risk factors: alcohol, smoking (mutates p53), dietary nitrosamines, aflatoxin, HPV infection, chronic esophagitis Nat'l history: epithelial dysplasia→ carcinoma in situ → invasive carcinoma Morphology: polypoid, exophytic masses, ulcerative/necrotic lesions (may cause TEF), diffusely infiltrative Micro: dysplastic squamous epithelium with keratin pearls. Cells have large nuclei and are unorganized Location: 50% middle third, 30% lower, 20% upper Complications: weight loss and debilitation, TEF and aspiration, hemorrhage and sepsis, metastasis (veins and lymph) and death. |
|
|
Barrett's esophagitis
|
Pathophys: caused by chronic inflammation (usually from GERD) resulting in adaptive metaplasia of intestinal mucosa into the esophagus
Macro: like pink normal mucosa with distal patches of red intestinal mucosa Micro: stratified squamous epithelium is replased by columnar epithelium with goblet cells (mucin) and villous formation - May progress to adenocarcinoma the metaplasia becomes invasive and no longer resemble a normal epithelium. This can then spread through veins and lymph |
|
|
Gastrointestinal stromal tumor (GIST)
|
Pathophys: Arise from interstitial cells of Cajal (component of the myenteric nerve plexus) and form masses that distend submucosa into the lumen and the serosal side into the abdominal cavity. Mucosa is not always replaced by tumor but can be.
Mutations: KIT receptor (CD117--codes for tyrosine kinase receptor which controls Ras activity), characteristic mutation results in proliferation of Cajal cells. Micro: large mass between muscle layers in the GI tract Location: most commonly in the stomach but also in the esophagus and intestine Complications: hemorrhage (usually presenting feature), invasion/compression of other organs. Cells are of mesenchyme origin Prognosis: determined by the size (large) and the number of mitotic bodies (many) |
|
|
Anatomical regions and functions of the stomach
|
Cardia: area just inferior to the GEj, mucous cells
Fundus + body: majority of the stomach. Contains parietal cells (secrete acid and intrinsic factor—B12 absorption) and Chief cells (secrete pepsinogen—protein digestion) Antrum: contain G cells (secrete gastrin) and D cells (secrete somatostatin), mucous cells |
|
|
Gastric acid secretion from parietal cells
|
Stimulation:
-histamine from enterochromaffin and mast cells - ACh from vagus nerve - gastrin secreted by G cells in the antrum. Also activates ECL cells Inhibition: -somatostatin: produced by D cells, directly inhibits parietal cells but also inhibits G cells. D cells activated by decreased pH - prostaglandins: prostaglandin E products |
|
|
Phases of gastric acid secretion
|
Cephalic: before food enters, prepares for digestion, stimulated by senses (sight, smell, taste) and thought thought → vagus nerve → enteric plexus → gastrin secretion (G cells) + histamine (ECL cells) + gastric acid (parietal cells) → ↑HCl
Gastric: stimulated by mechanical distention of the stomach → G cells (gastrin → ECL → H2) → ↑HCl Intestinal: minimum, very little acid production |
|
|
Peptic ulcer disease
|
= defect of the gastrointestinal surface of varing depth, affects stomach, duodenum, or esophagus
Pathogenesis: imbalance between acid/pepsin secretion (usually normal) and mucous layer (mucin, phospholipids, bicarb—prostaglandins bring blood to maintain it) Risk factors: Zollinger-Ellison syndrome (gastrin producing tumor), NSAIDS (inhibit PGs, direct injury. Esp: >60, steroid use, anticoag, prior history), H. Pylori, smoking Symptoms: dyspepsia (upper abdominal pain), fullness, bloating, nausea/distention, burning pain. Often in the middle of the night and/or 1-3hrs postrprandial, improves w/ eating, antacids Diagnosis: response to therapy, EGD if alarming symptoms, H. pylori test (IgG in new infection, urea breath test, stool antigen, biopsy urease test, histology, culture) Complications: bleeding (6-7% mortality, 80% resolves spontaneously), perforation (needs surgical correction, air under diaphragm), obstruction of outlet (from inflammation/scarring—succussion splash), penetration to adjacent organs (pancreas) |
|
|
H. pylori gastritis
|
- gram negative, curved, flagellated rod, likely oral-fecal transmission
- risk factors: age >60, lower socioeconomic class, developing countries, crowded living - specific to gastric mucosa (stomach or intestinal metaplastic areas), 80-95% of duodenal, 10-90% of gastric Pathogenesis: adheres to surface mucosa→ produces urease to protect against acid (locally alkaline), release virulence factors (VacA, CagA), mucinase and phospholipase → mucous degradation + cell damage. Also ↓D cells → ↑gastrin/HCl |
|
|
Gastritis
|
Acute: transient inflammatory process, many causes including alcohol, NSAIDS, ischemia, etc
Chronic: characterized by mucocal atrophy, imflammory infiltrate (lymphocytic) and intestinal metaplasia (globlet cell formation, villi development) - etiologies: H. pylori (assoc w/ PUD, adenocarcinoma, gastric lymphoma), autoimmune (10%, F>M, anti-parietal cells, pernicious anemia b/c no B12 absorbed), chronic alcohol abuse, radiation, post-surgical |
|
|
Gastric polyps
|
Hamartomatous: 80-90%
- non-neoplastic (no dysplasia or malignancy), may be regenerative in etiology (excess normal material) - associated w/ Peutz-Jegher syndrome (autosomal dominate, also has hyperpigmented macules on lips + oral mucosa) - associated w/ FAP (fundic gland polyps) Adenomatous polyps: 10% - neoplastic, dysplastic epithelium with malignant potential |
|
|
Gastric Adenocarcinoma: disease stats
|
Epi: Most common malignant gastric tumor, variable incidence (high in Japan, Chile, China, Russia, less in US, UK, Canada), a leading cause of cancer death worldwide. M>F 2:1, age >50
Risk factors: - environment: H. pylori, ↑nitrate diet, smoked/salty food, cigarettes, lack of fruits/veggies, low socioeconomic status - Host: chronic gastritis, intestinal metaplasia, adenomatous gastric polyps, Barrett’s esophagus, partial gastrectomy, Menetrier disease (thickened folds) - genetic: family hx, hereditary non-polyposis colon cancer syndrome (DNA mismatch repair), familial gastric cancer syndrome (E-cadherin mutation), familial autoimmune gastritis Etiology: mostly in antrum and lesser curvature (though cardia increasing b/c Barrett’s) Staging: T0 (carcinoma in situ), T1 (invasion of lamina propria or submucosa), T2 (invasion of muscularis propria or subserosa), T3 (penetration of the serosa), T4 (invasion of adjacent structures) Prognosis: tumor stage (based on depth). At 5yr, early 90%, late <15%. Intestinal type slightly better (presents earlier?). Lymph involvement: non >50% at 5yr, lymph metastasis <10% 5yr |
|
|
Types of gastric adenocarcinoma
|
Intestinal
- 50-60%, associated w/ environmental factors, usually older patients - usually develops from intestinal metaplasia (chronic gastritis) or adenomatous polyps - mass lesion, may be ulcerative/hemorrhagic or flat - micro: disorganized intestinal metaplasia (round gland formation). Cells are hyper chromatic w/ irregular nuclei Diffuse Type: - lacks environmental factors or precursor lesions, affects younger patients, M=F - associated w/ E-cadherin mutation (CDH1 gene) causeing cells to fall apart and loosely invade - “linitis plastic” morphology (leather bottle stomach)—stomach becomes thick walled/stiff due to infiltrate - Micro: cells falling apart, signet ring pattern (nuclei on the side) |
|
|
Gastric Lymphoma
|
- Most common extranodal lymphoma, primarily B-cell
Low grade: Mucosa-associated lymphoid tissue (MALT) - H. pylori in 62-77% of lesions, antibiotic therapy can be successful - clonal proliferation of small B lymphocytes due to chronic stimulus and inflammation - Prognosis: overall 65-90% 5yr, worse if t11:18 translocation High Grade: diffuse large B-cell lymphoma - arise denovo or in MALT lymphoma - treatment: surgery, chemo. Radiation - prognosis: 40-55% 5-year survival |
|
|
Gastric Carcinoids
|
- neuroendocrine tumors (often secrete gastrin), rare in the stomach
- can be associated with MEN-1 (multiple endocrine neoplasia type 1) - associated with Zollinger-Ellison syndrome |
|
|
Zollinger-Ellison Syndrome
|
- excess gastrin secretion associated with a carcinoid tumor (pancreas, stomach, small intestine)
- cause extensive peptic ulcer disease - Associated with MEN-1 (multiple endocrine neoplasia type 1) |
|
|
Menetrier Disease
|
Menetrier Disease - Disease of unknown etiology (endoscopic diagnosis), more common in males, 30-50yo
- defined by giant gastric folds and foveolar hyperplasia - causes protein losing gastropathy and hypochlorhydria, - increased risk of adenocarcinoma |
|
|
Intestinal malrotation
|
= abnormal intestinal orientation due to arrest of normal developmental rotation
4 types: - Non-rotation - incomplete rotation (may result in superior mesenteric artery obstruction) - reversed rotation (may lead to colonic obstruction) - anomalous fixation of the mesentery Presentation: - infancy (90% <1y), M>F, may be asymptomatic and not present until later or at all (less serious—occasional vomiting, pain, volvulus. Rarely enteropathy, pancreatitis, peritonitis, biliary obstruction, motility disorders, chylous ascites) - often affects the duodenum or upper jejunum resulting in intestinal obstruction (bilious emesis), peptic ulceration, malabsorption, infarct (hematemesis, melena, peritonitis), venous/lymph obstruction (steatorrhea) and generally pain/tenderness, distention, and shock. - Diagnosis: upper GI series shows distention of duodenum and abnormal duodenojejunal positioning, cecum on the left - Treatment: Ladd’s procedure |
|
|
Intestinal atresia
|
- debatable cause, possibly mesenteric vascular problems leading to loss of the intestinal lumen and obstruction
- Most commonly jejunal (50%) and duodenal (45%), more distal the more severe Presentation: bilious emesis (often first 15min of life), abdominal distention (first 2days), if untreated: constipation/impaction, weight loss, fretfulness, dehydration. Low grade atresia may present as abdominal pain and failure to thrive. Double Bubble visible of x-ray (suggests air build up in the duodenum) also dilated bowel proximal to obstruction Treatment: urgent surgery |
|
|
Congenital hypertrophic pyloric stenosis
|
= A cause of gastric outlet obstruction due to diffuse hypertrophy and hyperplasia of the smooth muscle of the proximal antrum in the first 1-2mo of life
- M>F, 1/300-900 live births. Etiology unknown Risks: family history, Turner’s syndrome, trisomy 18 - presentation: regurgitation at 2-3 weeks (confused w/ reflux), non-bilious vomiting 4-8wks, hyperperistalsis and abdominal mass, beak sign (?) on barium Treatment: splitting of muscularis via pyloromyotomy |
|
|
Hirshsprung disease
|
= congenital aganglionic megacolon: inability to relax in response to distention
- pathogenesis: loss of receptor tyrosine kinase RET (or other enteric plexus genes—endothelin, endothelin receptor) leading to abnormal migration of the neural crest cells distally or premature neural cell death so there is unopposed sympathetic tone from the Aurbach plexus and reduced NO. - 80% only distal rectum and sigmoid, severe cases effect whole colon (10-15%), colonic and small bowel (5-10%) Risk factors: Down’s syndrome (30%), other neurologic abnormalities, family history, M>F Presentation (from birth): inability to pass meconium, abdominal distention, constipation/impaction, difficulty passing flatus. Severe cases: enterocolitis (80% mortality), colonic rupture, fluid/electrolyte disturbances Diagnosis: suction biopsy (w/ stains for AChesterase), barium studies (aganglionic section is contracted, proximal is dilated) Treatment: surgical resection of affected segment. Complications: strictures, impaction, soiling, enterocolitis |
|
|
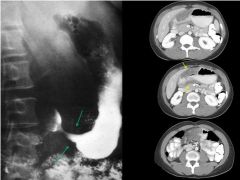
Gastric cancer
|
|
|
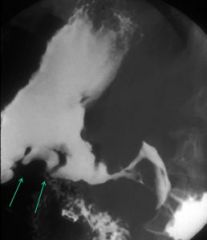
Malignant gastric ulcer
- may penetrate through the wall - encroaches the lumen - have nodular margins, typically irregular, not surrounded by smooth edema |
|
|
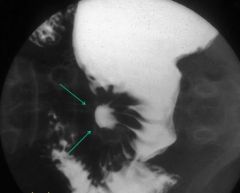
Benign gastric ulcer
- small collection of barium in stomach or duodenum - surrounded by edema (with folds) |
|
|
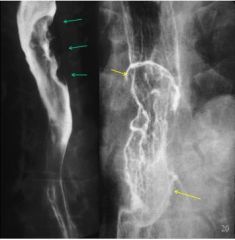
Esophageal carcinoma
- bitten appearance/indent appearance due to growth into the lumen. If severe/circumferential can have apple core appearance - Most common type: Historically squamous, now adenocarcinoma - Most common risk factors: Tobacco abuse, Alcohol abuse, Gastroesophageal reflux, Head and Neck cancer, Achalasia |
|
|
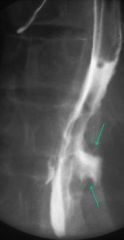
CMV esophagitis
- deep, penetrating ulcer |
|
|
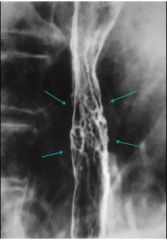
Barrett’s esophagitis
- nodular, irregular |
|
|
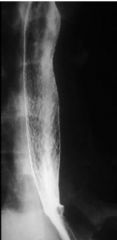
Reflux esophagitis (barium + effervescent granules)
- irregular, limited distention |
|
|
Antacids
|
Eg. Mylanta (aluminum hydroxide + magnesium hydroxide), Tums (calcium carbonate)
- treat reflux by reducing the acidity in the stomach by buffering acid (soluble salt product excreted) - generally not absorbed, act locally in the stomach Side effects: (mostly from excessive self-administration) - aluminum ions can complex with phosphate leading to hypophosphatemia - large increases in pH can impair absorption of acid-dependent food (require low-pH enzyme like pepsin) and drugs (require protonation) - hypercalcemia, due to use of calcium salts |
|
|
Mucosal protectants
|
Eg. Sucralfate (modified sucrose), colloidal bismuth compounds (peptobismol),
- treat reflux by protecting the mucosa from attack by acid and pepsin Mechanims: binds cell surface proteins (selectively binds damaged tissue) creating an artificial mucus layer, also binds pepsin and bile salts and suppresses growth of H. pylori - not absorbed, so few adverse effects Others: Misoprostol (prostaglandins, activate PG receptors on parietal cells stimulating mucus production) |
|
|
Histamine receptor antagonists
|
Eg. Cimetidine (also: Ranitidine, Famotidine, Nizatidine)
- treat reflux by reducing acid secretion Mechanism: selectively inhibits H2 receptors on basal surface of parietal cells preventing activation by H2/cAMP production, therefore reducing the overall activity of the H/K ATPase on the apical surface Side effects: (Cimetidine only) - inhibition of cytochrome p450: many food/drug interactions, requires careful monitoring - antiandrogenic effects: gynecomastia |
|
|
Proton pump inhibitors
|
Eg. Omeprazole, lansoprazole, rabeprazole, esomeprazole
- treat reflux/PUD by reducing acid secretion Mechanism: covalently binds parietal cell H/K ATPase, preventing acid secretion from all stimulatory pathways - prodrug activated by low pH, irreversible so long duration Side effects: few, despite being p450 substrate (side effects/tolerability vary with drug) Efficacy: resolves esophagitis and symptoms better than H2 antagonists, modest benefits of doubling doses |
|
|
Antibiotics therapy for PUD
|
- treat PUD caused by H. pylori infection
Always multi-drug therapy: 2 antibiotics+ antacid. Allows for synergistic healing by treating infection (complementary mechanisms) and reducing acid content First line: omeprazole + amoxicillin + clarithromycin Rescue: omeprazole + bismuth + tetracyclin + metronidazole (can also use levofloxacin, rifabutin) |
|
|
Signs and Symptoms of GERD
|
Common
- pyrosis (heartburn), dysphagia, regurgitation - often occur after high-fat or large meals, may be positional (better if sitting up) Severe (indicating EGD/imaging): - odynophagia, weight loss, GI bleeding, family history of GI cancer, anemia, advanced age Extra esophageal: - asthma, bronchitis, laryngitis, chronic cough, bloating, otitis media, frequent throat clearing, pneumonia, chronic sinusitis |
|
|
Nissen Fundopliocation
|
Indications: GERD with suboptimal response to PPI
Procedure: - treat loss of LES tone by wrapping fundus around the distal esophagus creating a 2cm zone of increase resistance to reflux - 5 steps: (1) diaphragm dissection and CNX preservation, (2) esophageal mobilization, (3) diaphragm closure, (4) division of short gastric vessels and posterior fundus, (5) fundoplication over esophageal dilator Prognosis: - generally good: 0.5-1% operative mortality, 3% require a redo, 5-10% have complications: gas-bloat syndrome (can’t burp, too tight), diarrhea (bruised CNX), chest pain, dysphagia, post-op flatulence. - Surgical failure is when: wrap slips down creating a pouch, wrap becomes undone, wrap is too tight causing esophageal distention, wrap herniates up over esophagus |
|
|
Types of hiatial hernias
|
- occur when the stomach protrudes through the diaphragm.
- risk factors: obesity and old age, which weaken musculofascial structures and allow for esophageal hiatus enlargement - symptoms are similar to GERD: epigastric discomfort, post-prandial bloating, dysphagia, anemia (from ulcerative bleeding). Severe complications include: bleeding, perforation, strangulation Types: (II-IV require always require surgical intervention d/t severe complications) - Sliding (I): (95%) cardia moves superiorly into the posterior mediastinum bringing GEJ through the diaphragm - Paraesophageal (II): fundus dislocates superiorly, GEJ normally placed - Mixed paraesophageal (III): both fundus and cardia dislocate superiorly bringing GEJ (most common paraesophageal type) - Paraesophageal type IV: when an additional organ (colon, spleen) herniates with the stomach |
|
|
Common operations for PUD
|
Indications: failure of medical management (now rare), bleeding/perforated ulcer
Vagotomy: remove CNS stimulation of acid secretion by selective excision of nerves of Latarget (to parietal cells). Preserves motility function Drainage procedures: pyloroplasty (revises pylorus), gastrojejunostomy (bypasses pylorus), anrectomy (resects pylorus. These improve drainage impaired by vagotomy Billroth I: anrectomy + vagotomy, remaining distal stomach and duodenum directly attached end-on Billroth II: same as above, except do a side-on anastomosis (leaving duodenal diverticulum) due to duodenal scarring |
|
|
Idiosyncratic drug toxicity
|
= drug toxicity that cannot be predicted based on dose, duration of exposure, or mechanism of drug action
- primarily an immune mediated phenomenon: drug-protein adducts acts as antigens driving lymphocytic attack of the tissue - may also be hypersensitivity response: associated with rash, fever, eosinophilia - Non-immune reactions: can result from aberrant drug metabolism or clearance, leading to accumulation of toxins and inhibition of critical cell processes - re-challenge can be dangerous even at low doses because reactions are specific/not-dose dependent - Drugs that idiosyncratic liver damage include: sulfonamides, amoxicillin-clauvanate, phenytoin, HIV meds |
|
|
Dose dependent drugs toxicity
|
- toxicity is predictable and reproducible because it is proportional to dose: drugs are toxic to everyone regardless of genotype/environment if taken in sufficient quantity
- ex: acetaminophen - treatment: cessation of drugs |
|
|
Factors that effect drug metabolism
|
Environmental/individual factors: Age, gender, pregnancy, disease state (esp liver), organ function (liver, GI, kidney), genetics, polypharmacy, diet, exercise, race, environmental toxins
P450 modifiers: - Inhibitors (increase drug circulation by reducing inactivating metabolism): grapefruit juice (only effects GI CYP3A4), ritonavir (used in HIV to increase potency of other drugs), acute alcohol, erythromycin, clarithromycin, fluconazole - activators (reduce drug circulation by increasing metabolism): St. John’s wart (CYP3A4), chonic alcohol, phenobarbital, phenytoin, rifampin, carbamazepine, steroids |
|
|
Phase II metabolism
|
= conjugation reactions, usually detoxifying, involving addition of polar functional group to phase I metabolites
- improves solubility, facilitating excretion Reactions: glucuronidation (most common), sulfation, aceylation, glutathione conjugation, amino acid conjugation, methylation |
|
|
Phase I metabolism
|
= biotransformation to increase polarity or activity by introducing/exposing functional groups, may produce toxic metabolites
- occurs in most tissues (GI epithelia, kidney, skin, lung), but primarily in the liver during first pass metabolism (improves solubility) - most phase I enzymes are located in the ER 3 types of reactions: oxidation (majority), reduction, hydrolysis Enzymes: cytochrome P450s, non-microsomal oxidases (MAO, ADH), reductases (azeoreuctases, nitroreductases) hydrolases (epoxide hydrolase, amidases, esterases (in intestinal epithelium, liver and serum)) |
|
|
Biotransformation of drugs
|
= chemical modification of compounds (nutrients, amino acids, toxins, drugs)
Main modifications: - increase polarity: facilitates renal exretion, transport in the blood (not bound to albumin) - change pharmacological/toxicologic activity: inactivation (usually) or activation |
|
|
Gross/micro findings of alcoholic cirrhosis
|
Macro: decreased liver size from fibrosis, diffuse nodularity (micro or macronodular), green-brown pigmentation from bile stasis
Micro: marked fibrosis surrounding viable tissue, residual inflammatory infiltrate surrounding hepatocytes of variable composition |
|
|
Pathogenesis/features of alcohol-related hepatis
|
Pathogenesis: alcohol is metabolized in the liver to acetaldehyde (alcohol DH) which is toxic to hepatocytes. Other byproducts are also toxic. Glutathione is used to reduce toxic free radicals, but it can be depleted (long term alcohol abuse). Excess amounts also increases release of specific endotoxins from the intestinal flora which can enter portal circulation causing mitochondrial and microtubule dysfunction.
Features: may progress to cirrhosis (but not a necessary precursor) due to prolonged exposure to fibrogenic cytokines. Injury may act synergistically with other conditions (viral hepatitis) Presentation: from acute weight loss, anorexia, malaise to severe cirrhosis (portal hypertension, jaundice, etc) |
|
|
Alcoholic hepatitis
|
= acute pathology typically following markedly excessive alcohol consumption. Typically resolves, but 10-20% mortality risk
- symptoms range: malaise, anorexia, fever, abdominal pain, tender liver, elevated hepatic enzymes w/ peripheral neutrophilia Macro: not too helpful, often accompanied by steatotic change, hepatomegaly, or fibrosis Micro: characterized by hepatocyte swelling and necrosis. Has eosinophilic intrahepatic inclusions (Mallory Bodies) of intermediate filament aggregates (cytokeratin). Inflammatory infiltrate (neutrophilic), often have fibrosis from longer term alcohol abuse |
|
|
Causes of hepatic steatosis
|
Alcohol
Drugs: abacavir & emtricitibine (antivirals) Pregnancy: especially 3rd trimester Non-alcoholic steatohepatitis: results from inflammatory and fatty change in patients with comorbidities (diabetes, obesity). Similar to metabolic syndrome, but diagnosis of exclusion Long term parenteral nutrition: important for ICU patients for whom liver damage could worsen condition |
|
|
Hepatic steatosis
|
= fatty accumulation of the liver. Can occur from alcohol within days or weeks. Rarely symptomatic but may have elevated enzymes and enlarged liver. Reversible, so resolve within several months when the precipitating factor is removed.
Pathogenesis: alcohol metabolism generates excess NADH which inhibits FA oxidation (so have decreased breakdown) alcohol also inhibits microtubule transport and lipid assembly, inhibiting lipid secretion. In the rest of the body fat catabolism increases, delivering more fat to the liver. Net: increase accumulation/synthesis, decreased breakdown/excretion Macro: large liver (up to 6kg), yellow greasy appearance Micro: intra )microvesicular)and extra-hepatocyte (macrovesicular) lipid accumulation (spherical droplets) - Alcohol typically results in microvascular lipid accumulation (but some overlap). In pure steatosis there is no necrosis or fibrosis |
|
|
Alcohol related hepatic pathologies
|
3 main types: Steatosis, Alcoholic hepatitis, Alcohol-realted cirrhosis
- there is continuum between these: progressive damage from toxic metabolites generated from alcohol result in steatosis and/or hepatitis (both reversible). Prolonged injury and inflammation (from either or both) result in collagen deposition and fibrosis, culminating in cirrhosis. |
|
|
3 main cause of cholestasis
|
= impairment of bile flow resulting in the retention of bile acids, bilirubin and cholesterol within the hepatic lobules. These accumulate in particular in the canaliculi resulting in distention (filled with bile). Over time obstruction ccan lead to portal edema, percholangitis, and cholestasis
3 main causes: drug induced, hepatitis, biliary obstruction |
|
|
Cirrhosis
|
= end-stage fibrosis of the liver
- results from chronic cell death, release of inflammatory cytokines, and collagen deposition - can be the end stage of many liver diseases including alcoholic liver, vitamin A poisoning (causes hyper osmotic state—chronic cell swelling), hemochromatosis, Wilson’s disease, chronic viral hepatitis, and others - results in pattern of islands of normal tissue between fibrotic bands, forming nodules (often green due to cholestasis). Liver shrinks as cells are replaced by collagen, and function declines with loss of cells, and cholestasis and portal hypertension can result |
|
|
Drugs associated with hepatitis
|
Isoniazid (for TB)
Methyldopa nitrofuran |
|
|
Irreversible liver damage
|
Necrosis
- results from progressive damage to cells (often beginning w/ swelling or steatosis) causing rapid cell death/lysis, expulsion of cellular products into the ETC space and phagocytosis by Kupffer cells. Then often triggers inflammatory response Apoptosis - controlled cell death (enzymatic, usually triggered by cytotoxic T-lymphocytes) resulting in the production of acidophil bodies (shrunken, eosinophilic cells due to the coagulation of cell products and nuclear material). Eventually remnants cleared by kupffer cells, not inflammatory |
|
|
Reversible liver cell injury
|
Cell swelling:
- can occur if ion transporters (esp Na/K ATPase) become non-functional (eg. hypoxia), membrane potential is lost allowing Na/H20 to enter the cell - In injury (eg. viral hepatitis) the cell membrane is progressively damaged losing barrier function, Na/H20 enter cell - result is dilution of cell contents: clear, vaculated cytoplasm, “ballooning generation” with normal nuclei Steatosis: “abnormal accumulation of TAGs within the cell” - classic change in alcoholic liver, also toxins (carbon tetrafluoride, acetominophen), diabetes assoc. w/ obesity, protein malnutrition - Results in large lipid vacuoles in cytoplasm on micro, and yellowing from large deposits on macro |
|
|
Biomarkers of hepatic syntheic capacity and function
|
Albumin: low not specific to liver. Reduced with poor nutrition, protein catabolism, nephrosis, protein losing enteropathy, burns, alcohol. Only indicative of chronic disease (T1/2 of 19-21 days)
Clotting factors/Prothrombin time/INR: measures conversion of prothombin to thrombin. Specific to liver because all factors (except VIII) are made there. But also sensitive to Vit K deficiency (ex: cholestatic patients), transfusions, congenital disease, liver disease, anticoagulants, vascular coagulation |
|
|
Bilirubin Synthesis and Metabolism
|
Heme oxygenase: breaks heme into more linear structure (biliverdin) which is then redcued to bilirubin. Oddities: catabolic pathway but uses NADPH, only process that produces CO
Insoluble bilirubin (solubilized by methanol/albumin) is then conjugated by UDP glucuronyltransferase Some is then converted to urobilinogen which can be oxidized and excreted by the kidneys as urobilin or conjugated bilirubin/urobilin can be excreted with the bile, and oxidized by bacteria to stercobilin which is excreted in stool |
|
|
Biomarkers of cholestasis
|
Alkaline phosphatase (ALP): <90U/L
- in bone, liver (bile canalculi), intestine, kidney, placenta but differentiate based on isoenzymes. - elevate after symptoms appear, indicates obstructive liver disease (hepatocellular carcinoma, bone disease, bile duct disease) γ-glutamyl transpeptidase (GGT): found on apical membranes of bile canaliculi cells, used w/ ALP to differentiate between hepatic vs. other ALP elevetions. Increased in heavy alcohol consumption, anti-epileptic medication 5’-nucleotidase (5’-NT): delayed release after injury (several days), not specific to liver (heart, brain, blood, vessels, pancreas) Bilirubin (<1.3 mg/dL): heme breakdown product, ciruculates in unconjugated/insoluble form (indirect) then conjugated by UDP glucuronyltransferase to soluble form (direct). Normally excreted by intestines/kidney as urobilinogen. Post-hepatic hyperbilirubinemia indicates deficiency in bile excretion/blockage (other pre/hepatic causes also) |
|
|
Biomarkers for hepatocellular injury
|
Alanine aminotransferase (ALT or SGPT) & Aspartate aminotransferase (AST or SGOT)
- normally contained in hepatocytes, released into blood after cell injury - normally function to transfer amino groups between glutamate and alanine - ALT more specific for liver disease than AST (also made by heart--MI, brain, pancreas, lung, WBC, RBC) - ALT is cytoplasmic (spills more easily into blood), AST is mitochondrial - AST/ALT <1 : acute liver disease - AST/ALT >1 : alcoholic hepatitis (often >1.5), acute Wilson’s disease, viral hepatitis w/ cirrhosis |
|
|
3 major types of hepatic dysfunction
|
Hepatic injury: may lead to hepatocyte degeneration and intracellular accumulations, hepatocyte necrosis/apoptosis, inflammation, regeneration, and fibrosis
- “A to I”: Autoimmune hepatitis, hepatitis B, hepatitis C, Drugs/toxins, Ethanol/hepatitis E (pregnancy), Fatty liver, Growths (tumors), Hemodynamic disorder (congenital heart failure/shock), Inborn errors (hemochromatosis, wilson’s disease, α1-antitrypsin deficiency) Cholestasis: occurs when bile cannot flow from the liver to the duodenum. - may be due to: cholestatic disease (primary biliary cirrhosis, primary sclerosing cholangitis, progressive familial intrahepatic cholestasis), bile duct obstruction (stone, tumor, stricture), infiltrative liver disease, space occupying lesions (tumor, abcess, lymphoma, granulomatous disease), or drugs (anabolic hormones, chlorpromazine, phenytoin) - presentation: jaundice, pruritis, skin xanthomas, malabsorption Reduced synthetic or functional capacity: can lead t o cirrhosis/end stage liver disease - most commonly caused by: alcohol, viral hepatitis, non-alcoholic fatty liver disease, hepatocellular cancer. Other causes: biliary cirrhosis, drugs (acetaminophen), hepatic congestion (cardiac cause), genetic disease (Wilson’s) |
|
|
Major function of the liver
|
Metabolism: urea cycle, gluconeogenesis, heme synthesis, cholesterol synthesis, drug metabolism
Regulates: blood glucose, fat storage Produces: bile, clotting factors, albumin & other blood proteins, hormones (IGF-1, thyroid, angiotensinogen, hepoidin), vitamins, essential immune system factors (complement system proteins) Detoxifies: alcohol, bilirubin, drugs Stores: glycogen, fat, minerals (iron) |
|
|
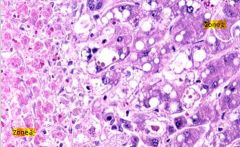
Acetominophen toxicity/overdose
- necrosis occurs worst in zone 3 where there is the lowest oxygen content, though all zones have fatty infiltrate - necrotic cells have coalescing fat vacuoles, shrunken appearance with abnormal nuclei |
|
|
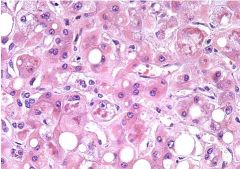
Irreversible changes due to chronic alcohol abuse
- progresses from steatosis to apoptosis (producing acidophil bodies) - Mallory bodies are also characteristic (though not specific): eosinophilic cytoplasmic inclusions of condensed aggregates of intermediate filaments (keratin) - grossly the liver becomes swollen due to fatty change, likely also associated with cirrhosis - inflammation is largely neutrophilic (segs) rather than lymphocytic. Over time leads to fibrosis and cirrhosis. |
|
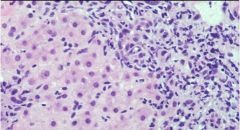
|
Liver injury from viral Hepatitis (pic is Hep B)
Agents: hepatitis A-E, CMV, EBV, yellow fever Pathogenesis: virus produces proteins with are expressed on the cell surface of MHC cells, which activate cytotoxic T lymphocytes, which surround lyse cells, releasing fibrogenic cytokines. Kupfer cells digest materials, but cytokines activate inflammatory and fibrotic changes Key: inflammation is largely lymphocytic Hep B: produce a lot of viral surface antigen resulting in ground glass appearing hepatocytes |
|
|
Acetominophen toxicity
|
Normal: 95% broken down to non-toxic metabolites, 5% broken down to NAPQI which must be detoxified by glutathione S-transferase (GSH) to mercaptopuric
Overdose: (most common cause of acute liver failure), 5% converted to NAPQI is sufficient to overwhelm glutathione stores and cause damage Chronic alcoholism: increases cytochrome P-450 activity, increasing relative amount of NAPQI formed, also uses up the GSH stores limiting neutralization N-acetylcysteine: antidote for overdose, augments glutathione reserve (directly binds NAPQI w/ GSH to enhance conjugation). Long term has antioxidant/anti-inflammatory effects, also facilitates liver perfusion as an inotrope and vasodilator. Oral forms not tolerated (low bioavailability, tastes gross, nausea vomiting). |
|
|
Hepatotoxic herbal remedies
|
– Niacin: like statins, it can elevate liver enzymes
– Vitamin A: elevates liver enzymes; only toxic at extremely high doses – Mushrooms: liver stigmata – Kava Kava – Black Cohosh: plant of the buttercup variety found in Georgia – Weight lifting and weight loss supplements – Senecio/crotalaria: Bush tea that may cause Sinusoidal Occlusion Syndrome – Chaparral- shrub, common in California – Germander- herb |
|
|
Acetominophen toxicity
|
3 Metabolic pathways:
- glucurondation: 40% - sulfation (sulfate conjugation): 20-40% - N-hydroxylation and rearrangement followed by GSH conjugation: <15%, CYP2E1/CYP1A2 metabolize to NAPQI which is irreversibly conjugated to the sulfhydryl groups of glutathione Each pathway results in a renally excreted product, pathway 3 intermediate NAPQI can cause hepatic damage |
|
|
Non-alcholic fatty liver disease
|
Epi: affects 20% of us population and rising.
Risk factors: obesity, diabetes, dyslipidemia (high TAG, low HDL) Pathogenesis: manifestation of metabolic syndrome. 2 hit hypothesis: 1. insulin resistance increases FA delivery to the liver. 2, increased FAs result in oxidative stress from beta-ox causing free radical formation, lipid peroxidation, and cellular damage Symptoms: usually asymptomatic, can have fatigue RUQ pain. PE: central obesity, hepatomegaly Lab: ALT>AST Treatment: underlying conditions |
|
|
Histopathology of NAFLD
|
Range of pathology from steatosis to cirrhosis. Indistinguishable from alcoholic changes (except by history).
Steatosis: fatty accumulation, occurs early in disease, no inflammation Steatohepatitis: fatty infiltration with varying degrees of inflammation and fibrosis. 20% will then develop cirrhosis Cirrhosis: fibrotic replacement of normal hepatic tissue |
|
|
Autoimmune Hepatitis
|
= self-perpetuating heptatic inflammation and fibrosis, diagnosis of exclusion
- 4x F>M - Pathogenesis: abberant immune response directed against hepatocytes (likely genetic predisposition plus environmental trigger) Presentation: fatigue, hepatomegaly, jaundice, other immune disorders (thyroid) Serology: ↑AST, ↑ALT, + ANA, SM antibody, liver-kidney microsomal-1 antibody Histology: interface hepatitis, hepatic rosettes, varying fibrosis Treatment: prednisone +/- azathioprine, 87% remission w/in 3 years of treatment |
|
|
Primary biliary cirrhosis
|
= disease characterized by progressive microscopic destruction of intrahepatic bile ducts
- F>M, associated w/ thyroid disorders, Sjrogren's, scleroderma, inflammatory arthritis Pathogenesis: aberrant immune response biliary epithelial cells (cholangiocytes) Presentation: fatigue, pruritis, jaundice, hepatomegaly, hyperpigmentation, xanthelasma Serology: ↑ALP, normal or mildly ↑ AST/ALT, ↑cholesterol, ↑IgM, fat soluble vitamin deficiency, Antimitochondrial antibody (diagnostic) Histology: bile duct destruction with inflammatory infiltrate Treatment: ursodeoxycholic acid (reduces mortality and transplant by 32%) |
|
|
Primary sclerosing cholangitis
|
= fibrosing inflammation of the intra and extra-hepatic bile ducts (affects large ducts)
- M>F, high association w/ IBD (esp UC) Presentation: cholangitis (abdominal pain, fever) Cholangiogram: multiple bile duct strictures w/ proximal dilation (due to fibrosis) Complications: irreversible damage to bile ducts leading to cholestasis, cirrhosis, liver failure, predisposes cholangiocarcinoma Treatment: transplantation |
|
|
Wilson's disease
|
= inherited disorder of copper metabolism
Pathogenesis: defect in hepatocyte Wilson ATPase, results in decreased copper excretion into the bile, results in increased systemic [copper] causing deposition and oxidative stress damage 5 clinical presentations: - hepatic: more typically younger patients, ranges from acute hepatic failure (5%) to mild chronic hepatitis, to cirrhosis and portal hypertension Neurologic: more typically older patients, rigidity, spasticity, tremor, ataxia, dysarthria Hematologic: hemolytic anemia from copper breakdown of RBCs, leads to bilirubin release and gall stones Psychiatric: typically older patients who have other symptoms, depression, psychosis, personality changes Opthalmologic: Kayser-Fleisher rings (copper deposition in periphery of cornea on slit lamp exam), sunflower cataracts (copper deposition on lens, do not affect vision, disappear with treatment) |
|
|
Signs of inborn errors of metabolism
|
- family history, especially if siblings are affected
- abnormal lab values - previously normal child begins to regress: ex. lysosomal storage diseases (loss of milestones or muscular or neurological degeneration), mucopolysaccharidosis causes progressive dymorphic changes in appearance - hepatomegaly: infant or adult, ex. Gaucher - hepatitis: alpha-1 anti-trypsin - acute or chronic liver failure: child or adult |
|
|
Genotype-phenotype correlation in inborn errors of metabolism
|
= genotype controls the severity of the disease
- range of presentations depending on the individual. Generally more severe mutation (deletion, missense mutation) the more severe disease (vs substitution) |
|
|
Alpha-1 Antitrypsin deficiency
|
= inborn metabolic deficiency of AAT which normally inhibits elastase form degrading elastin in the lung
- homozygous ZZ genotype has AAT <15%, leading to ephysema at a young age. - MZ genotype has AAT levels between 40-60% of normal, sufficient to keep lungs healthy unless combined with other risk factors (smoking) |
|
|
Basic pathological mechanisms for inherited metabolic diseases
|
All metabolic disease occur when there is block in a metabolic pathway resulting in either: 1, toxic build up of substrate or 2. lack of downstream products
Pathogenesis of 5 main diseases: GSD type 1: lack of glucose-6-phosphatase prevents conversion of G6P to glucose, causing hypoglycemia in infants and increased G6P (causing increased glycogen stores and hepatomegaly) Tyrosinemia: defective fumnarylacetoacetate hydrolase, leading to increase in toxic substrates (fumarylacetate and succinylacetone--inhibits conversion of 5-ALA to porphobilinogen causing porphyria Lysosomal storage disease: all involve toxic buildup of metabolite including sphingolipids (Gaucher's , Fabry's, and Neimann-Pick) AAT deficiency: lack of AAT allows elastase to degrade elasing in the lungs leading to COPD, also malformed AAT aggregates in the liver lead to inflammatory response and damage Hereditary hemochromatosis: defective HFE gene causing ferroportin dysfunction in enterocytes, hepatocytes, and macrophages leading to iron build up in the body |
|
|
Treatment strategies for metabolic diseases
|
1. Enzyme replacement therapy (may cause immune response b/c body has never seen correct enzyme before)
2. Reduction of toxic substrates 3. Supplementation of products Treatment for 5 main metabolic diseases: GSD type 1: provide product (glucose) with continuous feedings/cornstarch before bed, or liver transplant if diet fails Tyrosinemia: reduce toxic substrates with low protein diet and use NTBC to block pathway upstream of toxic fumarylacetate and succinylacetone LSDs: enzyme replacement/chaperone therapy to replace or help fold enzymes AAT deficiency: enzyme replacement given IV. Treats lung but not liver disease (malfolded protein accumulation) Hemochromatosis: reduce substrate with frequent phlebotomy (women less affected due to blood loss during menstruation) |
|
|
Family vs. population based screening for hemochromatosis
|
1/250 people are homozygous for C282Y HFE gene that causes most cases.Of these 70% have elevated ferritin and transferritin saturation, but <10% have organ damage
- Testing asymptomatic generally population would lead to identification of a large at-risk population but only a small number would develop disease. Also no effective treatment, only phlebotomy (doesn't improve morbidity/mortality) - In symptomatic families penetrance is still low, but serum ferritin is better predictor of outcome. If elevated w/o other cause proceed with genetic testing |
|
|
Benzimidazoles
|
Eg. Albendazole, mebendazole, triclabendazole
Mechanism: inhibits polymerization of tubulin and tubulin-dependent glucose uptake Activity: helminthes (ascaris, enterobius, hookworm, trichurius), microsporidia (encephalitozoon) |
|
|
Ivermectin
|
- derivative of avermectin, derived from soil mold (Streptomyces avermitilis)
Mechanism: binds to glutamate-gated chloride channels leading to paralysis and death Activity: GI/tissue dwelling nematodes (strongyloides, onchocerciasis), ectoparasites (scabies) |
|
|
Trimethoprim-sulfamethoxazole (TMP-SMX)
|
= Bactrim
Mechanism: synergistic inhibition of tetrahydrofolate synthesis—DHFR inhibitor (TMP), PABA inhibitor (SMX) Indications: broad spectrum antibacterial and antiprotozoal activity (isospora, cyclospora, toxoplasma) |
|
|
Nitazoxanide
|
= nitrothiazolyl-salicylamide dertivative
Mechanism: targets pyruvate-ferredoxin oxireductase (PFOR) enzyme-dependent election transfer reations Indications: giardia, cryptosporidia, other protozoa and anaerobic infections |
|
|
Nitroimidazoles
|
Eg. Metronidazole, tinidazole
Mechanism: reductive activation results in the formation of toxic free radicals causing DNA damage Toxicity: disulfiram effect (interaction with alcohol causing nausea, vomiting, flushing, tachycardia, SOB), serotonin syndrome Activity against: anerobic bacterial, protozoa Specific infections: giardia, trophs of E. hystolytica |
|
|
Hookworm
|
- primarily developing world/tropical/subtropical w/ poor sanitation, 700mil+ infected worldwide
Pathogenesis: acquired through the skin, migrate to lungs and small bowel, eat through the mucosa to feed off blood (.5 mL/day/worm) Symptoms: initially pneumonitis, ground itch. Significant worm burden can lead to significant anemia, albumin and other nutrient deficiency. Major contributor to malnourishment worldwide Tx: albendazole, mebendazole |
|
|
Trichuris trichuria
|
= “whipworm”
- primarily tropical disease affecting the colon via ingestion of eggs. No tissue migration - invade causing destruction of the mucosa and colitis-like symptoms - often asymptomatic or bloody diarrhea, colitis, rectal prolapse Tx: albendazole, mebendazole |
|
|
Enterobiasis vermicularis
|
= pinworm
- egg infection, day care centers, most common helminth infection in US Symptoms: anal itch (females migrate out of the rectum to lay eggs) or asymptomatic Dx: scotch tape on the bottom to look for eggs Tx: albendazole, mebendazole |
|
|
Ascaris lumbricoides
|
Epi: probably the most common infection worldwide (billion+), poor sanitation,
Spread by ingestion of eggs, then undergoes tissue migration (skin, lungs), adult worms in Jj and other locations Typically asymptomatic, except in high worm burden causing obstruction (b/c they’re so fucking huge). Acute presentations if they obstruct the biliary or pancreatic duct (cholecystitis, pancreatitis) Tx: albendazole, mebendazole |
|
|
Strongyloides stercoralis
|
Epid: tropical, sub-tropical, poor sanitation, southern US
Pathogenesis: infection via skin penetration of filariform larvae (soil, bare feet), tissue migration (to blood, lungs, trachea, GI tract), mature worms burrow in duodenum/jejunum, epithelial ulceration/trauma, larvae released to environment, autoinfection via circulation (hyperinfection syndrome) Presentation: chronic but usually asymptomatic (immune system keeps worm burden low), immune-suppression makes infection fulminant - skin (ground itch in initial infection, larva currens, uticaria), lung (pneumonitis, Loffler’s syndrome—wheezing, infiltrates, eosinophilia), GI (asymptomatic, abd pain, nausea, diarrhea, malabsorption), - hyper infection syndrome: fever, abd pain, wheezing, dyspnea, hemoptysis, sepsis (gram-negative) Dx: serology (screen before transplant), duodenal biopsy, stool studies are insensitive Tx: Ivermectin |
|
|
Apicomplexa
|
= intestinal protozoa, all cause self-limited watery diarrhea, or severe chronic illness w/ recurrence in immune suppressed pts (treat w/ immune reconstitution)
- Cryptosporidium parvum: round oocytes found in global surface water, contaminated water supplies (Milwaukee ’93), Dx: stool or biopsy w/ acid stain, Tx: nitazoxanide - Isospara belli (Cytoispora): “eye” shaped w/ 2 nuclei found mostly tropical, treated with trimethoprim-sulfa - Cyclospora cayetanensis: round oocytes found mostly tropical, imported produce, Dx: stool w/ acid stain, treated w/ trimethoprim-sulfa Also: microsporidia: infected globally by ingestion of spores, infect cells w/ sporoblast by extending polar tubule, Tx: albendazole, fumagilin |
|
|
Entamoeba histolytica
|
Class: amoeba, protozoa
Epid: developing world, immigrants, travelers Pathogenesis: invades the mucosa, mostly in the colon, ingested RBCs visible on micro Presentation: mostly asymptomatic, some cause bloody diarrhea (dysentery), inflammatory colitis, liver abscess (reddish brown—anchovy paste), RUQ pain, rarely brain/lung abcess Dx: stool microscopy, antigen immunoassay, serology Tx: trophs-metronidazole, cysts-paronomycin, iodoquinol |
|
|
Giardia lamblia (G. Intestinalis)
|
Class: flagellated intestinal protozoa
Epid: global via contaminated water sources (streams, ponds, resevoirs-beavers,etc), daycare centers Pathogenesis: cyst ingestion then excystation, trophozoites (look like balloons/droplets) attach to mucosa (duodenum and jejunum), non-invasive but can cause villous blunting, resulting in hypersecretion and malabsorption Presentation: mostly asymptomatic, acute disease (after 7-14d incubation period): steatorrhea, malaise, cramps, bloating, flatulence, nausea, weightloss, less common vomiting & fever. Can progress to chronic disease Dx: stool microscopy (ova and parasites), immunoassays (stool antigen), duodenal sampling (string test, aspirate, biopsy) Tx: metronidazole, tinidazole (forms toxic free radical damaging DNA, active against anaerobes, protozoa) |
|
|
Congential variants of the pancreaticobiliary tree
|
Must be cognizant of variants to prevent devascularization during surgery
- replaced left hepatic artery: comes off left gastric instead of proper hepatic - right hepatic artery branches off SMA below the pancreas instead of proper hepatic - accessory pancreatic duct abnormalities: extra “Duct of Santorini” connects straight to the duodenum at the minor papilla, bypassing the ampulla of vater (should be non-functional, whereas the duct of Wirsung caries the digestive enzymes). Some people may have larger accessory duct that carries most of the pancreatic juice - Cystic Duct anomaly: 18-23% of people, effect where the cystic joins the common bile duct (middle 75% vs distal 10% third), may spiral ant/post before insertion, low later insertion, common sheath w/ common duct, etc. In surgery effects where the duct can be clipped - Pancreas divisum: common abnormality, ventral/dorsal pancreatic ducts do not unite embryologically so majority of flow is through the minor papilla. Poor MIP + high flow may increase pancreatic duct pressure causing pancreatitis |
|
|
Surgical management of acute pancreatitis
|
- Peustow-Gillespy procedure: for dilated pancreatic duct (>7cm) w/o mass, attaches the tail to a limb of the intestine allowing flow from that end. 30% still have pain (from head or uncinate process)
- Beger procedure: resection of most of head and body, sleeve left to supply blood to duodenum - Frey procedure: coring out of pancreatic head then pancreaticojejunostomy |
|
|
Complications of pancreaticobiliary surgery
|
Biliary: 50% have complications:
- encroaching vessels (may be unresectable or require venous patch), bile lead or biliary fistula (may resolve or require radiological/surgical intervention), biliary stricture (obstructs bile flow, surg/rad correction), pancreatic fistula (pancreas to other organs leaking secretions), pancreatic necrosis (accidental vessel resection), Diabetes (from removal of tissues), exocrine insuffiency |
|
|
Surgical procedures for resection of pancreatic tumors
|
Head of the pancreas (also intestinal, pancreatobiliary): Whipple procedure (pancreaticoduodenectomy)
- removal of gastric antrum, 1st and 2nd portions of the duodenum, head of pancreas, common bile duct, gallbladder. Reconnected through gastro/hepatico/pancreatico-jejunostomies Neck: often not resectable because of entanglement w/ the SMA, otherwise Whipple Tail: distal pancreatectomy (open or laproscopic). Pancreas dissected longitudinally, vessels cut away or divided, distal pancreas w/ accompanying veins removed as a unit |
|
|
Surgerical procedures for resection of cholangiocarcinomas
|
Intrahepatic duct (5-10%): require resection
Hilar bile duct (60-70%, “Klatsin”): resected along the biliary tree, reconstructed w/ hepatojejunostomy and removal of liver on the affected side. Classified with the Bisthmuth classification (type IV, both sides + lymph, unresectable) Distal Bile ducts (20-30%): resected with whipple procedure Gallbladder tumors: laparotic cholecystectomy or open surgical (better if larger or invasive to the liver). Not resectable if there is vascular invasion |
|
|
Pancreatic Islet cell tumors
|
= rare pancreatic endocrine tumors (2% of pancreatic tumors)
- majority are functional (produce expected hormone), 15-35% are clinically silent - difficult to predict the biologic behavior (malignant/benign): all look the same histologically (homogenous cells, stain/serum for horomones), bloody macroscopically (large vascular supply) - associated with Multiple Endocrine Neoplasia Type-1 syndrome (pituitary, parathyroid, pancreatic lesions) Types: - Insulinoma (47%): body and tail, usually benign, causes shakes/sweats, uncontrolled hypoglycemia (Whipple’s triad) - gastrinoma (28%): most common malignant, body and tail, causes hyperglycemia (diabetes) and necrolytic migratory erythema (peeling skinn - VIPoma (vasoactive intestinal peptide, 14%): causes watery diarrhea, hypokalemia, achlorhydria - glucagonoma (10%): 2/3 malignant, causes peptic and small bowel ulcers (Zollinger-Ellison syndrome) - somatostatinoma (1%): causes diabetes, steatorrhea, hypochlorhydria, cholelithiasis |
|
|
Pancreatic Acinar cell tumors
|
= rare tumors of the enzyme secreting cells of the pancreas
- can result in lipase, trypsin, and amylase secretion: these digest the pancreas resulting in painful nodules of fat necrosis (can be used to differentiate from pancreatitis) - Poor prognosis, though slightly better than ductal adenocarinoma |
|
|
Clinical features of pancreatic adenocarcinoma
|
Presentation: epigastric pain (can be asymptomatic until late stage, especially in tail), unexplained weight loss, jaundice (may be painless), migratory thrombophlebitis (Trousseau’s sign), elevated CA 19-9
Gross: firm, fibrotic, can occur in the background of chronic pancreatitis, can look like fibrotic pancreatitis on imaging Histo: infiltrative islands of cells coursing through fibrotic tissues, duct forming (not necessarily good ones) Distribution: head (80-90%), body (5-10%), tail (10-15%) Metastasis: (<20% confined to pancreas) lymph nodes, liver, peritoneum. May also invade the spleen Prognosis: <5% at 5yrs (mostly because discovered so late) Staging: T1 (limited to pancreas, <2cm), T2 (limited to pancreas, >2cm), T3 (beyond pancreas, not in celiac axis or SMA), T4 (beyond pancreas and involves celiac axis and/or SMA) |
|
|
PanIN to pancreatic adenocarcinoma progression
|
PanIN 1: proliferation
- Her-2neu, K-ras (activation of oncogene, most common mutation) PanIN 2: proliferation and dysplasia and some dysmorphia - p16 (inactivated tumor suppressor, majority of ductal ACs also familial melanoma) PanIN 3: adenocarcinoma insitu, large nuclei, crowed cells - p53 (inactivated suppressor), DPC3, BRCA2 |
|
|
Epidemiological and Etiological factors of pancreatic adenocarcinoma
|
Epid: most common malignant neoplasm of the pancreas (85-90%), effects older patients: 60-80yrs, 4th leading cause of cancer death
Etiologic factors: - Environmental factors (loose association): smoking, petroleum products, lack of fruits/veggies, alcohol abuse (pancreatitis) - Host factors: chronic pancreatitis, diabetes, pancreatic intraepithelial neoplasia (PanIN, precursor lesion) - genetic factors: 1st degree relative, HNPCC, BRCA2 , familial dysplastic nevus/melanoma syndrome, Peutz-Jegher’s syndrome, hereditary pancreatitis |
|
|
Pancreatoblastoma
|
- malignant tumor of infancy/childhood, rare cases in adults
- epithelial and mesenchymal elements - better prognosis than ductal adenocarcinoma, adults have worse prognosis than children |
|
|
Cystic pancreatic neoplasm
|
Serous cystic neoplasm: mainly in body and tail
- generally benign (serous cystadenoma), malignant behavior less common (serous cystadenocarcinoma) - F> M Mucinous cystic neoplasm: - less predictable biologic behavior, F>M Solid pseudopapillary tumor: - most have indolent growth (benign pancreatic mass), some are aggressive/metastatic - almost exclusively young females |
|
|
Adenocarcinoma of the gallbladder
|
= most common malignant tumor of the biliary tract
Epid: in the US highest among Hispanics, native Alaskans, and native americans; worldwide highest in Indian women. Generally F>M, patients tend to have gallstones (60-90%) Risk factors: trauma & inflammation due: to chronic cholelithiasis/cystitis (in US), pus forming or parasitic diseases of the biliary tree (Asia) Presentation: similar to cholelithiasis—asymptomatic or severe abdominal pain, nausea, and vomiting. Complications: erosion through the GB into the liver or peritoneum Gross: thick walled GB, shaggy material growing in the fundus Micro: gland forming, cells have lost natural polarity |
|
|
Metastatic carcinomas of the liver
|
= more common than primary neoplasm
- commonly from the colon (via portal circulation) Appearance: multiple focal lesions in the setting of normal liver (cirrhotic liver usually not ameniable to metastases) |
|
|
Cholangiocarcioma
|
= malignant tumor arising from the extra or intra-hepatic biliary ducts or the hilum (origin of common Hepatic duct – Klatskin tumor) or more distally in biliary tree
Nat’l hist: can cause obstruction and dilation of the biliary tract causing jaundice and hepatic obstruction. May metastasize to peritoneal surfaces causing massive ascites Risk Factors: primary sclerosing cholangitis, cysts of the biliary tree, thoratrast (old radiologic dye), chonic infection with liver fluke (Chinese liver flu—Opisthorchis sinensis) Micro: from duct-like structures |
|
|
Hepatocellular carcinoma
|
= malignant neoplasm of hepatocytes
Presentation: silent hepatomegaly (otherwise asymptomatic), rapid hepatomegaly in patients w/ cirrhosis plus worsening of ascites and pain, elevation of α-fetoprotein (50%) Risk factors: chronic infection with Hep B (or Hep C), chronic alcoholism (esp. w/ cirrhosis), aflatoxin exposure (from Aspergillus on grains), hemochromatosis Micro: wide colums of cells, many mitotic figures, dyspmorphic and enlarged nuclei, may produce bile, vascular invasion, may see mallory bodies in alcoholic liver Macro: green and nodular from cirrhosis plus yellow , smoother tumor area, may protrude from surface Prognosis: good if patient has single tumor <2cm, poor otherwise (~7mo if comorbid cachexia, variceal bleeding, liver failure) |
|
|
Hepatic Adenoma
|
= benign tumor of the hepatocytes
Macro: can reach 30cm in diameter, hemorrhagic Micro: pretty unremarkable hepatocytes (pale cytoplasm—glycogen), no kupffer cells, look for mitotic figures/vascular invasion (to distinguish from carcinoma) Epid: usually women of childbearing age who used OCPs (less common now that OCPs less potent), also men on anabolic steroids Presentation: either asymptomatic or RUQ fullness/mass +/- pain, acutely from hemorrhage Complications: significant hemorrhage (leading to hypotension, shock, death, if perforated in to peritoneum) esp. subcapsular adenomas. Also small risk for developing carcinoma Tx: surgical resection |
|
|
Focal Nodular Hyperplasia
|
= benign mass of hyperplastic degenerative hepatocytes, second most common liver tumor
Pathogenesis: usually forms in response to local vascular injury or congenital arteriovenous malformation Macro: characteristic central stellate scar Micro: lobular proliferation of hepatocytes with ductular proliferation and malformed vessels within the fibrous scar Epid: most commonly in women of reproductive age Presentation: most are asymptomatic, usually discover incidentally on scan Tx: these are benign and remain so, often resected b/c difficult to differentiate from hepatic adenoma |
|
|
Hepatic cavernous Hemangioma
|
= benign neoplasm of blood vessels in the liver, most common primary tumor
Macro: red/blue spongy mass Micro: composed of large endothelial cell-lined vascular channels and stroma, with RBC and thombosi inside Epid: F>M, may enlarge in women on OCPs (but not causative) Presentation: <4cm usually asymptomatic, >4cm RUQ pain/discomfort , fullness Dx: most pick up incidentally on scans, NO needle biopsy (can bleed chronically) Tx: surgical excision, alcohol embolization |
|
|
Gilbert’s Syndrome
|
- hereditary condition of impaired bilirubin metabolism
- pathogenesis: reduce activity of UDP glucuronyl transferase, resulting in elevated unconjugated/indirect bilirubin - presentation: recurrent jaundice in response to stress, otherwise asymptomatic, M>F - management: none/stress relief. ERCP is unnecessary |
|
|
Distal malabsorption due to resection
|
- resection of the ileum >100 cm causes uncompensated loss of bile acids (not sufficient resorption) resulting in impaired fat digestion (presents with steatorrhea). Require chronic B12 supplementation
- resection of ileum <100 cm: liver can compensate for reduced bile reuptake so patients have diarrhea with mild/no steatorrhea. These patients will not respond to low-fat diet, but will respond to cholestyramine. Require chronic B12 supplementation. |
|
|
Bile
|
- composed of: bile acids, cholesterol, phospholipids, bile pigments (bilirubin), ions, water
- continually produced and secreted by hepatocytes, concentrated and stored in the gallbladder, secreted into the duodenum in response to chyme - emulsifies fats to enable absorption - bile acids reabsorbed in the ileum (95%) and returned to the liver by the portal circulation. Reduces demand to synthesize new bile acids |
|
|
Somatostatin
|
- peptide hormone released by the D-cells in the gastric/duodenal mucosa and the pancreatic islet
- stimulated by low pH, inhibited by vagus stimulation - INHIBITS gastric acid and pepsinogen secretion, pancreatic and small intestine fluid secretion, gallbladder contraction, and release of insulin and glucagon |
|
|
Secretin
|
- protein hormone secreted by the S-cells of the duodenum upon stimulation by acid and fatty acids in the duodenum
- increases pancreatic enzyme and HCO3 secretion - decreases gastric acid secretion - alkalinizes the duodenum to facilitated pancreatic enzyme function |
|
|
Cholecystokinin (CCK)
|
- peptide hormone
- produced in the I-cells of the duodenum and jejunum upon stimulus by small peptides, amino acids, and fatty acids - Increases pancreatic enzyme and HCO3 secretion - Stimulates gallbladder contraction and relaxes the sphincter of Oddi - inhibits gastric emptying: slows stomach so intestine can digest fats - stimulates satiety via CNS receptors |
|
|
Celiac disease
|
= gluten (gliadin) hypersensitivity
- characterized by flattening and atrophy of the mucosal villi (mucosal scalloping/mosaic pattern), lymphocytic and plasma cell infiltration of the lamina propria - Results in malabsorption: iron, folate, fat-soluble vitamins Presentation: diarrhea, weightloss, iron-deficiency anemia, low serum albumin, (Irish descent) |
|
|
Fat, protein, carb digestion and absorption
|
Carbs:
- degraded by amylases in the saliva and pancreatic juice. Then by sucrose, maltase, and lactase in the intestine. - Glucose, galactose then absorbed by Na/X cotransport, and fructose by facilitated diffusion Fats: - emulsified in the duodenum and broken into micelles (TAGs, cholesterol esters, phospholipids) - TAGS: digested into monoglyceride and FA by lipases - Cholesterol esters: digested into cholesterol and FA’s by cholesterol ester hydrolase - Phospholipids: digested into lysolecithin and FA’s by phospholipase A2 - End products absorbed by enterocytes and repackaged into chylomicrons Protein: - stomach: digested into amino acids and oligopeptides by pepsin - small intestine: digested in to di-/tri- peptides by pancreatic enzymes (trypsin, chymotrypsin, elastase, carboxypeptidase A/B) and brush border peptidases (oligopeptides). - Di and tripeptides absorbed in the small intestine by Na/X cotransporters |
|
|
Conjugated vs. unconjugated bilirubin elevation in hepatic disease
|
↑Unconjugated: pre or intrahepatic obstruction of bile processing
- Gilbert’s Syndrome: UDPGT deficiency/decreased activity - Crigler-Najjar syndrome: UDGT deficiency - Physiologic jaundice of the newborn: increased breakdown of erythrocytes, low UDPGT activity - Hepatitis ↑Conjugated: intra or post hepatic obstruction of excretion - Dubin-Johnson syndrome: defective human canalicular multispecific organic anion transporter (cMOAT). Impairs non-bile salt anion transport - Bile duct obstruction - Hepatitis |
|
|
Gallstone Ileus + Mirizzi Syndrome
|
Gallstone Ileus
= impaction of a gallstone in the ileum after passing though biliary-enteric fistula - stones in the gallbladder can promote fistula formation - stones get stuck at the anatomical narrowing of the bowel in the terminal ileum Dx: CT Tx: high morbidity/mortality (delayed dx), surgical removal Mirizzi Syndrome: - obstruction of the common hepatic duct by a gall stone in the cystic duct - Tx: surgery +/- ERCP |
|
|
Choledocholithiasis
|
= intermittent obstruction of the common bile duct by passed or de novo formation of stones
Presentation: often asymptomatic, intermittent pain (depending on the rate/degree of obstruction & bacterial contamination), mild hyperbilirubinemia, pruritis, elevated ALP, transient spikes in AST/ALT when stone passes to duodenum Dx: abd US only finds 50% of stones, use endoscopic (98% sn/sp), MRCP, ERCP Tx: high chance of complication, treat all w/ ERCO + cholecystectomy (unless high risk for surgery) |
|
|
Biliary Colic
|
= intermittent obstruction of the cystic by gallstone (+/- inflammation of the gallbladder)
Presentation: intermittent RUQ/epigastric (+radiation) pain <1hr may be worse with meals, usually normal PE/Labs b/c of intermittent symptoms Dx: US of gallbladder to find stones Tx: don’t treat asymptomatic stones (30% no further attack in 2yrs), otherwise elective cholecystectomy |
|
|
Cholelithiasis pathogenesis
|
= formation of stones in the gallbladder or bile tract
Pathogenesis: - cholesterol supersaturation: when there is significantly more cholesterol than bile salts, it precipitate out as crystals - accelerated nucleation: increased aggregation of crystals into larger microstones. Controlled by surface tension, pressure, temp, protein content - gallbladder hypomobility: bile in the gallbladder promotes the formation of stones. Increased stasis during TPN, pregnancy, rapid weight loss, octreotide Types of stones: cholesterol (80%), black pigment (hemolysis, cirrhosis, pancreatitis), brown pigment (biliary infection—stricture, duodenal diverticuli) Risk Factors: fat, female, forty, fertile. Also rapid weight loss, diabetes, TPN, elevated TAGs, drugs (Fibrates, OCPs, ceftriaxone, octreotide), disease of the terminal ileum (salts not absorbed, higher cholesterol content), cirrhosis, genetics (Scandinavians, Pima Indians) Complications: biliary colic, acute cholecystitis, choledocholithiasis, acute cholangitis, gallstone ileus, Mirizzi syndrome |
|
|
Chronic pancreatitis
|
= chronic inflammation resulting in permanent damage to the pancreas. Characterized by chronic abdominal pain, progressive loss of pancreatic endocrine/exocrine function
Pathogenesis: mainly chronic cystic duct obstruction Etiologies: repeat acute pancreatitis, alcohol (70%), idiopathic (20%), others (10%: hereditary, CF, pancreas divisum, autoimmune) Presentation: abdominal pain (painless ~15%), weight loss/anorexia, nausea/vomiting, malabsorption (diarrhea, steatorrhea, azotorrhea), pancreatic diabetes Complications: pseudocyts, ascites, pancreatic fistula, splenic vein thrombosis Dx: ERCP, endoscopic ultrasound, MR, US, AXR, CT (calcification), secretin/CCK, Bentiromide, pancreaolauryl test, fecal elastase, fecal chemotrypsin, serum trypsin, fecal fat, serum glucose (NOT amylase/lipase) Management: pain control (avoid alcohol, analgesics, uncoated pancreatic enzymes, celiac plexus block), endoscopic repair (stones, stricture), surgery, control diarrhea (coated enzyme therapy) |
|
|
Acute Pancreatitis
|
= acute, self-limited inflammation of the pancreas, characterized by acute abdominal pain, elevated pancreatic enzymes
Pathogenesis: premature activation of zymogens inside the pancreas leading to 1. Protease damage of acinar cells, 2. enzymatic fat necrosis by phospho/lipases, 3. Elastase damage of vessel walls causing hemorrhage, 4. Activation of circulating enzymes leading to SIRS Complications: SIRS, ARDS, shock, multiorgan failure, DIC, pleural effusion, acute renal failure, myocardial depression, metabolic dysfunction (hypokalemia, hyperlipidemia, hyperglycemia), pancreatic ischemia, necrosis and apoptosis (from generation of inflammatory mediators), pancreatic pseudocyts, infection/sepsis Etiologies: (GET SMASHED) gallstones, ethanol, trauma (seatbelts), steroids, mumps/malignancy, autoimmune, scorpion stings/structural, hypercalcemia/lipidemia, ERCP, Drugs (dideoxyinosine, 6-MP/azathioprine, diuretics)…also inherited (CF), vascular, idiopathic Presentation: acute abdominal pain, nausea/vomiting, tachycardia (hypovolemia), fever, guarding, loss of bowel sounds, jaundice (uncommon) Diagnosis: 3x increase in amylase & lipase, Ranson criteria, ERCP, CT w/ contrast Management: supportive care, surgery if indicated (infection, necrosis) |
|
|
Presentation of pancreatic cancer
|
- varies based on tumor size, location, stage
- common symptoms (often none/non-specific): abdominal pain (vague or dull/deep), weight loss, jaundice, back pain, vomiting, indigestion, pruritis, diabetes. - other symptoms: nausea, malaise, weakness, anorexia, diarrhea, early satiety, bruising, steatorrhea - if biliary obstruction results from tumor (head of pancreas): dark urine, pale stool, pruritis Dx: CT, double duct sign (biliary &pancreatic dilation) Treatment: chemo, radiation, Whipple’s procedure |
|
|
Cholangitis
|
= inflammation of the common bile duct due to fixed obstruction (impacted stones, stricture, neoplasm). Usually causes bacteremia (GI bugs)
Presentation: - early: fever w/o jaundice - late: Charcot’s triad (fever, jaundice, RUQ abdominal pain) or Raynaud’s pentad (Charcot’s + shock + altered mental status), prominent leukocytosis, elevated bilirubin, elevated ALP, often positive blood cultures PE: mild RUQ tenderness, hepatomegaly Dx: Charcot’s, EUS/MRCP, CT Treatment: IV fluids, broad spectrum antibiotics, urgent decompression/ERCP (if worsening symptoms), subsequent cholecystectomy |
|
|
Cholecystitis
|
= inflammation of the gallbladder, commonly associated with cystic duct obstruction (stones or biliary sludge)
Pathogenesis: bile stasis→ inflammatory mediators → mucosal damage of the gall bladder → ischemia, ulceration, edema, infection Presentation: - Fat, female, forty, fertile (multiple children) - symptoms: RUQ or epigastric pain >6hrs (episodic, often postprandial/night, radiation to rt scapula), anorexia, fever, nausea +/- vomiting, change in mental status (elderly) PE: murphy’s sign (inspirational arrest during deep palpation of gallbladder), RUQ tenderness, palpable GB (30%), mild jaundice, leukocytosis, mild AST/ALT/ALP elevation Dx: US to find stone/block, thickened gallbladder w/ surrounding fluid, HIDA scan (GB motility), CT Treatment: 83% resolve w/o complications in a week, cholecystectomy (preferred), NPO, fluid, antibiotics, cholecystostomy (stoma created when surgery not indicated) |
|
|
Complications of liver transplant
|
Short term complications:
- primary non-function: graft doesn’t work, rare today b/c of UW solution which stabilizes hepatocytes - delayed graft function: a form of acute failure - acute cellular rejection: incidence 50% in first year, but lower after with good medication compliance (generally immunologically mature organ so rejection not as much an issue) - hepatic artery thrombosis/stenosis: from the anastomosis, can also effect bile ducts - portal vein stenosis/thrombosis - bile duct stricture/leak - Disease recurrence: eg. Cirrhosis due to viral infection Long term: - hypertension, dyslipidemia, diabetes, obesity, skin cancer, other cancers, renal impairment, symptomatic fractures |
|
|
Medications post liver transplant
|
Cyclosporine and tacrolimus
- calcineurin inhibitor: inhibits the production of IL-2 turning off T-cells Sirolimus (rapamycin): - mTOR inihibtor, modifies target of rapamycin (TOR) to tostop IL-2 from activating T cells MMF/Azathioprine: cell cycle inhibition Steroids |
|
|
MELD score
|
= Model for End-Stage Liver Disease
- ranking score for liver transplant based on total bilirubing, INR, and creatine levels - range 6-40, transplant eligible >15 (risk of disease > risk of transplantation) or MELD< 15 with complications refractory to medical management. MELD >18 shows significant benefit of transplant, at emory average is mid 20’s |
|
|
Diseases treated with liver transplant
|
Chronic cirrhosis: (cirrhosis is the top reason for transplant, usually seen in patients w/ end-stage liver disease)
- Hep C (#1 US), Alcohol related cirrhosis (#2), Hepatitis B (#1 in asia), NASH ( fastest growing cause), PBC, PSC, autoimmune hepatitis, alpha 1 AT, hemochromatosis, wilson’s disease Fulminant liver failure: - acetaminophen toxicity, Hep A/B/E, acute alcohol, drug toxicity, AFLP, Budd-Chiari syndrome, idiopathic causes Other causes: - familial amyloidotic polyneuropathy, polycystic liver disease (assoc. w/ PCKD), hepatocellular carcinoma (2nd fastest growing) and cholangiocarcinoma, biliary etresia (#1 pediatric cause), inborn errors of metabolism |
|
|
Metabolic abnormalities due to liver failure
|
- hypoglycemia: decreased hepatic gluconeogenesis and glyconeogenesis, associated w/ severe failure and poor prognosis
- Lactic acidosis: impaired hepatic metabolism of lactate (Cori cycle) - hyponatremia: due to excess total body water Treatment: glucose infusion, dialysis |
|
|
Hematologic abnormalities in chronicl liver failure
|
- elevated prothrombin time: decreased factor synthesis, most reliable indicator of hepatic synthetic function
- decreased hepatic fibrinogen synthesis - thrombocytopenia: secondary to splenomegaly and splenic sequestration - Disseminated intravascular coagulation: if co-existent infection occurs Treatment: - prevent cold coagulopathy: FFP/recombinant Factor VIIa/Platelets/fibrinogen - Vit K to replete nutritional deficiency/malabsorption from cholestatic disease |
|
|
Hepatorenal syndrome
|
= functional renal failure in the presence of decompensated cirrhosis (serum Cr > 1.5g/dL or CrCl <40mL/min)
Mechanism: neurohormonal response to splanchnic vasodilation causing renal vasoconstriction and decreased perfusion Character: no improvement in renal function after fluid challenge and withdrawl of diuretic therapy, fractional excretion of Na (FeNa) <1, urine Na <10mmol/L Diagnosis: based on exclusion (eg: acute tubular necrosis, pre-renal azotemia, post-renal etiology) Treatment: splanchnic vasoconstrictors (vasopressin), plasma volume expansion (albumin), liver assist devices, liver transplant (if dialysis >6wk, liver/kidney transplant) Prognosis: poor in the absence of liver transplant (creatine defines severity of disease, transplant list ranking) |
|
|
Ascites and Sponateous Bacterial Peritonitis from portal hypertension
|
= peritoneal fluid accumulation secondary to renal fluid retention and portal HTN.
- SAAG > 1.1 (serum album to ascites gradient) b/c capillary membrane intact Complications: - massive ascites can cause restrictive lung physiology and abdominal compartment syndrome - ascitic fluid predisposes spontaneous bacterial peritonitis: peritoneal infection (ANC >250/cc) in the absence of perforated viscus. Organisms: gram +/-, fungi Treatment: - chronic ascites: Na restriction, diuretics, paracentesis, TIPS - SBP: antibiotics (emergent then prophylactic), albumin (reduce renal dysfunction), paracentesis |
|
|
GI variceal hemorrhage in portal hypertension
|
- varices form due to collaterals between the portal and systemic circulation
- rupture of the submucosal gastroesophageal, gastric, or rectal varices and result in potentially fatal hemorrhage Diagnosis: endoscopy Treatment: - Pharm: splanchnic vasoconstrictors (octreotide, terlipressin), non-specific β-blocker (decrease CO and splanchnic vasoconstriction), empiric antibiotics (treat bacterial translocation) - endoscopic band ligation (esophageal), balloon tamponade, surgical to acutely decrease portal pressures (TIPS, portocaval shunt, esophageal devascularlization) |
|
|
Hepatic hydrothorax
|
= accumulation of ascitic fluid into the pleural space, typically right hemithorax
Symptoms: dyspnea, hypoxemia Diagnosis: abnormal lung exam, CXR Comlications: respiratory failure, infection (spontaneous bacterial empyema) Therapy: thoracentesis, diuretics, TIPS |
|
|
Portopulmonary Hypertension
|
= form of pulmonary arterial hypertension histologically identical to idiopathic PH, can lead to cor pulmonale
Pathophysiology: unknown (some factors coming from the liver), likely vascular injury/inflammation from serum factors abnormally persisting in hepatic outflow Signs/symptoms: right heart failure, ascites, and peripheral edema Diagnosis: transthoracic Echo, rt heart cath for pulmonary pressures Treatment: (not immediately reversible with liver transplant) - Acute RV failure: diuresis, vasoactive agents - Liver transplant candidates: initiate prostaglandin therapy to decrease pressures, continue after transplant |
|
|
Hepatopulmonary syndrome
|
= abnormal dilation of the pulmonary vasculature due to systemic vascular changes from cirrhotic liver (in absence of lung lesions)
Symptoms: Platypnea (upright SOB), orthodeoxia (upright desaturation) Mechanism: dilation of basal pulmonary vasculature (mechanism unknown) impedes diffusion mediated transfer of O2 Diagnosis: cardiac bubble study (detection of microbubbles in left heart after 3-6 cardiac cycles (vs. 1-2 in ASD, 0 in normal) Treatment: supplemental O2 (treats hypoxemia), liver transplant (complete reversal in months to year) |
|
|
Pulmonary complications of liver failure
|
Hepatopulmonary syndrome: abnormal dilation of pulmonary vasculature causing hypoxemia (in the absence or radiologic lesion)
Portopulmonary Hypertension: pulmonary HTN causing cor pulmonale Hepatic Hydrothorax: transudative pleural effusion, typically right sided, due to passage of ascitic fluid into the thoracic cavity |
|
|
Cardiovascular derangements due to decompensated liver failure
|
- splanchnic vasodilation results in decreased systemic blood volume and sympathetic activation causing hyperdynamic circulation
- PE: tachycardia, low mean arterial pressure, wide pulse pressure, warm extremities (peripheral vasodilation) - Cirrhotic cardiomyopathy: systolic/diastolic dysfunction (high output failure), conduction abnormalities, impaired β-adrenergic receptor function |
|
|
Hepatic encephalopathy
|
= neurologic and psychiatric dysfunction in the presence of decompensated cirrhosis
Grading: clinically from Stage I (mild confusion) to stage IV (coma) Mechanism: conversion of excess ammonia (not eliminated by dysfunctional liver) to glutamine, induces astrocyte swelling and altered neuro-transmission HE in acute vs. chronic liver failure differs in disease manifestation, therapy and prognosis: in chronic liver failure gradual up-regulation of ammonia fixing systems (muscle) prevents ammonia bolus to brain (as occurs during acute decompenstation) Precipitating Factors: Infection, dehydration, GI bleed Markers: serum ammonia not sensitive for diagnosis but can be used to gauge treatment response Treatment: (typically reversible w/ treatment) - elimination of nitrogenous sources from GI tract to decrease NH3 - medication: lactulose, rifaximin, metronidazole (eliminate N producing gut flora) |
|
|
Extra-hepatic manifestations of liver disease
|
Hepatic encephalopathy
Variceal hemorrhage Hepatorenal syndrome Hepatopulmonary syndrome Ascites & spontaneous bacterial peritonitis Coagulopathy Hemodynamic abnormalities |
|
|
Clinical signs of cirrhosis
|
Stigmata of chronic liver disease: muscle wasting, vascular spider angiomas, impaired estrogen metabolism (palmar erythema, gynecomastia, hypogonadism in males), abnormal labs (AST/ALT >1, low albumin, INR >1.3, high bilirubin)
Palpable left lobe of the liver Small liver small Splenomegally (and thrombocytopenia as spleen sequesters platelets) Signs of decompensation: jaundice, ascites, asterixis |
|
|
Pathologic changes in the systemic circulation during portal hypertension
|
Vasodilation in the splanchnic artery (due to increased pressure) decreases effective arterial volume in the rest of the body
- Difficult to maintain systemic blood pressure: increased cardiac output leading to high output failure - Hypovolemia triggers neurohormonal responses (renin-angiotensin, sympathetic NS, ADH): results in salt/water retention causing ascites - Neurohormonal response causes renal vasoconstriction and decreased renal perfusion, when combined w/ high CO causes hepatorenal syndrome (leading to rapid renal failure) |
|
|
Pathophysiology of increasd splanchnic pressure in portal hypertension
|
2 main changes increase pressure:
Increased Resistance: structural changes (fibrosis, regenerative nodules), active vasoconstriction (decreased NO, increased vasoconstrictors) - hepatitis causes normally quiescent stellate cells to produce collagen, constricting sinusoidal spaces Increased flow: Arterial vasodilation in splanchnic circulation, mediated by NO production. (better target for therapy) - Increased superior/inferior mesenteric venous pressure (mild portal HTN): causes edema, decreased bacterial gut clearance, increased bacterial translocation to the bowel wall which enter the mesenteric nodes triggering NO production |
|
|
Etiologies of portal hypertension
|
Pre-hepatic: portal or splenic vein thrombosis (rare, often caused by polycythemia vera or factor V Leiden mutation), splenomegaly (increases splenic vein flow)
Intra-hepatic/sinusoidal: cirrhosis (most common), schistosomiasis, steatosis, sarcoidosis, veno-occlusive disease (obstruction of small veins: chemo, BM transplant injury), nodal regenerative hyperplasia Post-hepatic: Budd-chiari syndrome (hepatic ourflow obstruction), right heart failure, constrictive pericarditis |
|
|
Treatment for constipation
|
Dietary modification: 8 glasses fluid/day, bulk fiber, lactulose
Stool softeners: work in a fraction of patients Emollient: acts as lubricant to facilitate movement Osmotic laxatives: magnesium citrate, polyethylene glycol (for colonoscopy preps, only 1/wk) Stimulant laxatives: (antraquinones, castor oil) irritate the colonic mucosa causing strong contractions (and usually cramping pain) Chloride channel activators: (Luboprostone) secrete Cl to increase fluid Treat or manage underlying secondary causes of constipation |
|
|
Diganostic tests for constipation
|
History: make sure patient meets criteria, other symptoms, PMH
PE: descent and ascent of the perineum, anal reflex, check for anal prolapse, digital rectal exam (check relaxation of the puborectalis and perineal descent), vaginal exam Labs: CBS, glucose, Cr, Ca, TSH Procedures: colonoscopy or Ba enema (for structural causes) Motility tests: ballon expulsion, anorectal manometry (pressure in the rectum, check for appropriate relaxation), colonic transit study (patient swallows 24 radio opaque rings, scan on 5th day only 5-20% should remain, look for areas of stagnation) |
|
|
Constipation of the elderly
|
10 D’s
Drugs (Side effects) Defecatory dysfunction Degenerative disease Dementia Decreased mobility Dependence on others for assistance Decreased privacy Dehydration Depression |
|
|
Etiologies of constipation
|
Primary:
Lack of fiber/fluids IBS Abnormal motility: slow transit constipation (colonic inertia-muscles don’t work well/less nerve input, decreased high propagated contractions), uncoordinated motor activity (resulting functional barrier resistance to normal transit) Pelvic floor dysfunction: pelvic floor dysnergia, ano-rectal dysfunction Secondary: Disease of the colon: stricture, cancer, anal fissure, proctitis Metabolic disturbances: hypercalcemia , hyporthyroidism, diabetes mellitus, heavy metal intoxication Neurological lesions: - peripheral: hirschsprung’s disease, Chagas, pseudoobstruction, DM - central: multiple sclerosis, parkinsonism, spinal cord lesions (sacral nerve transection/injury, meningomyelocele, low spinal anesthesia: hypomotility, dilation, decreased rectal tone and sensation, stasis in distal colon) Drugs: analgesics, anticholinergics, antidepressants, antipsychotics, cations (iron, aluminum), neutrally active agents (opiates, antihypertensives, Ca channel blockers, ganglion blockers, vinca alkaloids, 5HT3 antagonists) |
|
|
Constipation
|
= when has 2 or more symptoms for >2weeks in the past 6mo:
- <3 defecations/week - straining w/ quarter of bowel movements - lumpy/hard stools - sensation of incomplete evacuation - manual maneuvers to facilitate defecation (digital evacuation) |
|
|
Differential for IBD
|
- UC vs CD
- Bacterial infection: M. tuberculosis, Yersinia, Salmonella spp, Shigella spp, C. jejuni, O157:H7, C. difficile - Viral infections: CMV, HSV - Neoplasia: carcinoid, colon CA, lymphoma - Functional: IBS, anorexia, sexual abuse - Vascular: ischemia, radiation - Drug related: NSAID - Inflammation: Eosinophilic gastroenteritis |
|
|
IBD extra-intestinal manifestations
|
Arthritis/arthralgias: 22%
Failure to thrive: often seen in children long before the present with abdominal pain and diarrhea Skin: erythema nodosum (tender nodular lesions on extensor surfaces of extremities), pyoderma gangrenosum (necrotizing inflammatory lesion often seen on shoulders and lips Joints: peripheral arthritis that is monoarticular, asymmetrical, usually in large joints. No synovial destruction of subcutaneous nodules, Rf factor negative. Usually parallels activity Eyes: uveitis/iridocyclitis (occurs in 90% of patients), episcleritis (inflammation of skin covering sclera) Hepatic: primary sclerosing cholangitis (patients with controlled disease may still have abnormal LFTs) Bones: sacroileitis (sclerosed bone and tissue in SI joint, independent of disease control), ankylosing spondylitis |
|
|
IBD therapy summary
|
UC:
- induction: steroids, 5-ASA, biologics - maintenance: 5-ASA, 6-MP/AZA, biologic CD: - induction: steroids, antibiotics, biologics - maintenance: antibiotics, 6-MP/AZA, biologics Mechanisms: 5-ASA: reduce prostaglandins 6-MP/AZA: inihibit purine synthesis in B and T lymphocytes Biologics: antibodies against cytokines and adhesion molecules |
|
|
IBD pathophysiology
|
- Etiologic hypothesis: combination of persistent infection, defective mucosal integrity, dysregulated immune response and dysbiosis (derangement of bacteria) contribute to development of disease
- disruption of intestinal epithelial barrier function (mucus, PGE2, IgA, keratinocyte growth factor, transforming growth factor β, intestinal trefoil factor, tight junctions, defensins) - dysregulated immune response with loss of tolerance (IL-1β, IL-23, TNFα, INFγ) IBD genetics: all activate common IL-12/Th17/IL-17 pathway, polygenic - UC: positive family history 10-15%, epithelial barrier dysfunction genes - CD: positive family history 15-25%, correlates severity and distribution, autophagy genes (CARD15/NOD-2) |
|
|
Celiac disease
|
= celiac sprue, non-tropical sprue, gluten enteropathy
- genetic disorder affecting primarily people of European descent. Associated w/ other autoimmune dz’s (SLE, RA, thyroid, dermatitis herpetiformis) - autoimmune reaction to gliaden fraction of gluten, primarily effects the duodenum Mechanism: tTG alters gluten peptides, encounters HLADQ2 or HLADQ8 forms complexes that activate T cells and mediate autoimmune destruction of villae (villous flattening and crypt hyperplasia) Presentation: may be asymptomatic, may have childhood presentation. Classically after ingestion of gluten: diarrhea, gas, bloating, weight loss, failure to thrive. Also: malabsorption, Fe deficiency, osteopenia, other nutrient deficiencies Dx: serum TTG, or anti-endomysial Ab, HLA typing (DQ2/8), small bowel biopsy Tx: strict gluten free diet, steroids if refractory, nutritional/vitamin replacement |
|
|
Causes of malabsorption
|
Carbs: (most common causing chronic diarrhea)
- normal: starch, sucrose, lactose broken down by amylase and disaccharidases (cellulose not digested) - Primary mal: enzyme deficiencies (lactase), transporter defects (Na/Glucose dependent transport) - secondary mal: reduce surface area (celiac, resection), deficient pancreatic enzymes (biliary obstruction) Consequences: flatus (unabsorbed carbs fermented in colon) Fat: - normally: most absorbed in proximal 2/3 of jejunum, broken to FA’s, active transport across membrane - malabsorption: shortened bowel, IBD, infections, celiac, chronic pancreatitis (deficient pancreatic enzymes), decrease bile salts (meds, biliary obstruction) Consequences: deficiencies in fat soluble vitamins (ADEK) and minerals (Fe, Ca, Mg), steatorrhea Protein: - normally: pepsin then trypsin and otherbreaks into AA or small peptides - malabsorption: transporter defects, pancreatic enzyme insufficiency, shortened bowel Consequences: protein deficiency, azotorrhea |
|
|
Differentiating between types of diarrhea
|
History:
- causes of maldigestion/malabsorption: previous surgery (resection), travel/exposures (giardia, amoeba), medications (antacids, cholestyramine, antibiotics), radiation therapy - causes of secretory diarrhea: diet high in poorly absorbed carbs (fructose) - Family history: lactose intolerance, celiac, crohn’s, CF PE: - weight loss (chronic malabsorption), steatorrhea w/ large volume (fat malabsorption), edema/muscle atrophy/hypoalbuminemia (protein malabsorption), watery diarrhea/flatulence/acidic stools (carb malabsorption), microcytic anemia/low Fe in bone (Ca malabsorption), macrocytic anemia (folic acid malabsorption) Serum: check TTG or EMA for celiac, vitamin (ADEK—fat soluble) or electrolyte deficiencies Stool: steatorrhea, azotorrhea Endoscopy: look for mucosal damage (scalloping in celiac) |
|
|
Categories of Diarrhea
|
Watery osmotic: occurs when unabsorbed (osmotically active) solute remains in the lumen, causing water to stay with it
- can be due to overload of poorly absorbed ions (Mg—antacids, oral supplements, laxatives; phosphate), lactose intolerance, ingestion of sugar-free products (not digested), ingestion of osmotically active substances (fiber), ingestion of trace elements in large doses - commonly occurs with either maldigestion or malabsorption of normal nutrients: congenital (transporter or enzyme deficiencies), acquired (muscosal damage, post infectious), progressive (enzyme deficiencies—biliary obstruction, CF), iatrogenic (post resection or diversion) Watery secretory: occurs when excessive stool water is secreted into the lumen due to the presence of unabsorbed electrolytes or excess secretory stimulation - Causes: infections (esp toxin producing), reduction in absorptive surface (Celiac), inflammation of the intestinal mucosa (IBD), secretagogues (agents that cause secretion of another substance), bile acid malabsorption (medications, ileal abnormalities—inflammation, resection) Inflammatory: IBD, ischemic, infectious, radiation induced Fatty: maldigestion, malabsorption (eg: mucosal disease, celiac, crohn’s, short bowel) |
|
|
Diarrhea
|
Normal stool: 75% water (100ml/day), 25% solids
Diarrhea = frequent defecation with a change in consistency (eg. Water, fatty, bloody, azotorrhea) and amount (>200g stool/day) Acute <4 weeks (usually self-limited), chronic (>4 weeks) |
|
|
Treatment for colorectal cancer
|
Site depedent:
- surgery: segmental resection - chemo: FOLFOX (folic acid analog + 5-fluoracil/antimetabolite + oxaliplatin/DNA crosslinker), FOLFIRI (folic acid + 5-FU +irinotecan/topoisomerase-1 inhibitor) - radiation: rectal cancer only |
|
|
Guidelines for colorectal cancer screening
|
75% of the population is at risk for sporadic CRC, the rest are at elevated risk due to genetic or pre-existing conditions (familial, HNPCC, FAP, IBD, MAP, hamartomas). Screening is based on risk group
For asymptomatic adults >50yo - tests that detect adenomatous polyps or cancer: flex sig/5yrs, or colonoscopy/10yrs, or Ba enema/5yrs (not good), CT colonography/5yrs - tests that detect cancer (<50% sensitivity): annual guiac, or annual fecal immunochemical test, stool DNA (interval incertain) |
|
|
Hereditary colorectal cancer syndromes
|
FAP:
- autosomal dominant disorder (1/8000, 100% penetrance) caused by germline mutation in APC gene - characterized by multiple adenomas by teenage years, and development of carcinoma by 4th decade - extracolonic manifestations: fundic gland polyps, desmoid tumors, osteoma of the mandible, congential hypertrophy of the retinal pigment epithelium (CHRPE, poor-man’s diagnostic test) HNPCC: - autosomal dominant disorder (1/1000) caused by germline mutation in a DNA mismatch repair gene (MMR) leading to microsatellite instability phenotype - 50-80% will develop CRC, w/ average onset in 5th decade - increased risk for extracolonic malignancies: endometrial, gastric, ovarian, uterine, (not breast unlike BRCA2) - Diagnosis w/ Amsterdam criteria (3-2-1 rule): 3+ relatives w/ CRC (at least 1 first degree), 2+ generations of CRC, 1+ case <50yrs Others: APC I1207 (attenuated from of FAP), MYH-associated polyposis, hamartomatous polyposis (Peutz-Jegher, Juvenile Polyposis) |
|
|
Pathways of genetic muations in colon cancer
|
Chomosomal instability: accumulation of mutations over time causing progressive dysplasia, and leading to carcinoma
Β-catenin or APC (dysplasia) → COX-2 (early adenoma) → K-ras (intermed. adenoma) → DPC4/SMAD4 (late adenoma) → p53 (carcinoma) Microsatellite instability pathway: involves mutations associated with DNA repair mechanism (MSH2, MLH1, MSH6, etc) |
|
|
Colorectal cancer general
|
Epid: 4th most common cancer, 2nd most common cause of cancer death, mortality of 1/3 (50% in 2002), lifetime risk is 6% in US.
Distribution: 40% right, 60% left colon, most often metastasizes to the liver Staging: based on extent of infiltration. TNM and/or Duke criteria (mucosa, submucosa/A, muscularis propria/B, serosa/C, metastasis/D) |
|
|
Neoplastiv vs non-neoplastic colorectal polyps
|
Non-neoplastic:
- hyperplastic: usually small, result from decreased cell shedding leading to the accumulation of normal tissue (well differentiated glands, mature crypts, serrated appearance) -inflammatory - lymphoid - hamatomatous: involves epithelium, lamina propria, and muscularis mucosa. Arise in Peutz-Jegher’s and juvenile polyposis syndromes Neoplastic: - adenoma: (Benign but progress, 5-7yrs) -- serrated: mixed hyperplastic and adenoma -- tubular: 80% of all neoplastic polyps, most common adenoma, often small pedunculated, dysplastic cell originate in the crypt epithelium and move up, develop crowded cytoplasms and nuclei -- tubulovillous -- villous: usually large and sessile, have villous architecture but have hypercellularlity, hyperchromasia, more basophilic, pleomorphic nuclei, and reduced goblet cells - carcinoma: (malignant) non-invasive (carcinoma in-situ), invasive(glands infiltrate beyond the epithelium) |
|
|
Epidemiology of colorectal cancer
|
4th most common cancer in men and women (143K new cases/year)
2nd most common cause of cancer death (9%, 52K) Aggressive screening programs have reduced deathrate after diagnosis from 50% to 33% in the last 10 years Lifetime risk of developing CRC in the US is 6% |
|
|
Osmotic vs. secretory diarrhea
|
Osmotic = occurs as a result of insult to the gut mucosa that impairs absorptive ability. Unabsorbed osmolytes accumulate in the lumen, increasing the osmolarity and preventing free diffusion of water into the enterocytes. Stool therefore has a higher water content
Secretory = occurs in response to overstimulation of secretory pathways by pathogens. Eg. Cholera toxin (activates adenyl cyclase to increase Cl transporter activity), E. coli heat-labile enterotoxin (activates guanylyl cyclase), VIP secreting tumors (activate adenyl cyclase) |
|
|
Water and electrolyte absorption/secretion
|
Absorption: occurs in the villous cells (tall columnar w/ microvilli and abundant golgi/ER)
- majority of water absorbed in the small intestine (6700mL), 1500 in the colon, 100mL excreted in feces. - in the small intestine electrolytes and nutrients are actively transported (transporters, pumps, channels) while water is passively absorbed (paracellular or cellular pathway), usually regulated by Na (used as a cotransporter for peptides and glucose—exploited in rehydration therapy) Secretion: occurs in the crypt cells (short cuboidal) - water secretion mainly governed by choride secretion via the Na/K/Cl2 co-transporter on the serosal surface (K and Na can thebe reabsorbed by transporters—Na/K exchanger, K channel). The mucosal surface then has a Cl channel (CFTR, requires VIP for expression, PKA for activation) allowing for Cl/H20 secretion (this is broken in CF) Balance: maintained by endocrine (bile, guanylin promote Cl secretion) and neuronal controls (ACh, VIP—control Cl secretion), especially on secretion. Also influence by the speed of passage/motility in the gut. |
|
|
Acute colonic pseudo-obstruction
|
= syndrome with clinical features of obstruction without any mechanical obstruction. Likely a disruption of parasympathetic tone OR a increase in sympathetic with decrease in parasympathetic
Epid: patients with stroke, MI, peritonitis, sepsis, after surgical procedures Clinical features: severe abdominal distention with the absence of stool or gas passage Dx: AbXR (dilation of ALL segments of colon) Tx: correct revesible potential causes, neostigmine, endoscopic decompression, percutaneous cecostomy, surgical decompression Prognosis: 0-32% mortality |
|
|
Ileus
|
= failure of normal intestinal motility in the absence of an obstructing lesion
Etiologies: abdominal or retroperitoneal surgery, inflammation, metabobolic, neurogenic, medications Clinical features: poorly localized abdominal pain, abd distention, nausea, vomiting, obstipation, quiet or absent bowel sounds, no fever/tachycardia or peritoneal signs Dx: AbXR (presence of gas in stomach, small intestine, colon), CT to confirm Tx: limit PO, fluid resuscitation (fluids, electrolytes, NG tube) |
|
|
Large bowel obstruction
|
Etiologies:
- most common: Malignancy (>50%), volvulus (10-15%--twisting of colon on vascular pedicle, mostly sigmoid or cecum), stricture secondary to diverticulitis (<10%) - less common: Crohn’s, endometriosis, intussusception, extrinsic factors, fecal impaction Clinical Features: abd pain (periumbilical or hypogastric), abd distention, diarrhea or obstipation (depending on degree and location) - Benign colonic structures: change in stool caliber - Malignant colonic strictures: change in stool caliber, blood in stool/iron deficient anemia, weakness, weight loss, anorexia, vomiting (late finding), often insidious at onset - Colonic volvulus: acute abdominal distention (66%), abdominal pain, nausea, vomiting, constipation, shock (10%), average 3days of symptoms before presentation Dx: AbXR (dilated colon proximal to obstruction), CT, barium studies, colonoscopy has limited clinical value Tx: fluid resuscitation (fluids, electrolytes, NG), benign (operative management), malignant (colonic stents, operative management), sigmoid volvulus (decompression, rectal tube, elective resection), cecal volvulus (fix to wall, create fistula, electively resect) |
|
|
Small bowel obstruction
|
Etiologies:
- most common: post-operative intra-abdominal adhesions (50-75%), hernias (25%), neoplasms (5-10%) - less common: congenital atresia/stenosis, inflammatory (IBD, ischemia, post-anastomotic, diverticulitis, radiation, drugs), intussusception, foreign bodies (gallstones, feces, bezoars), volvulus, carcinomatosis, endometriosis, abscess Clinical features (magnitude depends on site, degree, duration): abd pain (acute onset), vomiting, obstipation, abd distention PE: high pitched bowel sounds, increased BS w/ periods of quiet, abd tenderness and guarding (suggests strangulation), systemic (indicated dehydration): tachycardia, tachypnea, altered mental status, oliguria , hypotension Lab: ↑WBC, abn electrolytes, renal failure Rad: AXR (dilated bowel loops), CT (dilated loops, incarcerated hernia), contrast studies Tx: IV fluids, foley, correct electrolytes, NG tube (decompress and minimize distention), then proceed to laparotomy (+ antibiotics if peritonitis) or spontaneous resolution |
|
|
Post-diarrheal infection complications
|
- Chronic diarrhea: lactase deficiency, small bowel bacterial overgrowth, malabsorption syndromes
- Initial presentation or exacerbation of IBD - Irritable bowel syndrome - Gullain-Barré syndrome—Campylobacter jejuni - Reactive arthritis: particularly Campylobacter, invasive pathogens - Hemolytic uremic syndrome (HUS): STEC (E. coli O157 and others, Shigella), children <5 yr |
|
|
Epidemiology of Acute infectious diarrhea
|
- Traveler’s Diarrhea: ETEC
- Cruise ships: Norovirus - Children <2 years: Rotavirus - Hospitalized or recent antibiotics: C. difficile - Chittlin (chitterlings) exposure: Yersinia (may have appendicitis-like presentation), Winter Holidays - Undercooked hamburger: E. coliOl5J or other STEC - Church picnic: Staphylococcal food poisoning - Travel to Russia, US National Parks: Giardia, Cryptosporidia |
|
|
Norovirus
|
= virus of the family Caliciviridae (small, round ssRNA-positive sense), formerly Norwalk-like viruses. Most common cause of foodborne outbreaks in the US
Clinical: 10-51hr incubation, sudden onset of N/V, diarrhea, lasting 1-2 days Transmission: fecal-oral, aerosol-vomitus, fomites, contaminated food (infected handler), contaminated surfaces (cruise ships), direct contact (family members, 30% secondary case rate) Tx: supportive therapy only |
|
|
Rotavirus
|
= non- enveloped, icosahedral, triple-layered particles with dsRNA in 11 segments (Reoviridae family)
Epid: in US: primary cause of acute GIitis in young children (nearly every child by 5) few deaths. Globally: 600-850k deaths/year, 1child/20min Transmission: fecal-oral and possible airborne, winter seasonality Presentation: vomiting, fever, followed by watery diarrhea causing dehydration and electrolyte abnormalities Dx: antigen assays, RT-PCR, culture Tx: rehydration, NO antivirals Prevention: live, oral vaccines for infants |
|
|
Food poisoning
|
Pre-formed toxins: incubation 1-4hrs
- symptoms: N/V, diarrhea - organisms: S. aureus (potato salad, cream pastries, mayo), bacillus cereus (fried rice, spores) Pre-formed or quickly produced toxins: incubation 8-16hrs - symptoms: abdominal cramps, diarrhea - organisms: clostridium perfringens, B. cereus Microbial contamination of food products: incubation >16hrs - symptoms: watery or inflammatory diarrhea - organisms: V. cholera (shellfish), ETEC (misc), EHEC (ground beef, milk, raw produce, juice), salmonella (poultry, eggs, beef, dairy, peanuts), campylobacter (poultry, milk), shigella, vibrio parahaemolyticus (mollusks, crustaceans), norovirus (N/V, diarrhea) |
|
|
Clostridium difficile
|
= toxigenic, spore-forming, anaerobic gram positive rod
- causes antibiotic associated diarrhea/colitis, often in hospital (recent rise in non-healthcare settings) Pathogenesis: colonization w/ C. diff, disruption of the normal flora (by antibiotics), toxin elaboration (A, B, binary) mediates diarrhea and colitis Clinical: varies from mild, watery diarrhea to bloody dysentery; classically results in pseudomembranous colitis Dx: positive C. dif toxin A/B +/- culture Complications: shock, toxic megacolon, perforation. Sepsis, death |
|
|
Salmonella
|
= gram negative rod, non-spore forming, motile with peritrichous flagella (family enterobacteriaceae), typically in sewage contaminated foods( eggs, poultry, produce)
- 7 sub species (of one species), most human disease in subspecies 1. Isolates sterotyped by somatic O, surface Vi, and flagellar H antigens (used in species designation) Types of infection: Enteric fever (typhoidal): salmonella typhi, choleraesuis, paratyphi A/B. Usually infection from human sewage Enterocolitis (non-typhoidal): salmonella enteritidis and typhimurium (and >1400 others). Most are foodborne diseases, use animal as a reservoir (farm animals, pets, contaminated foods) - decreased acidity in the stomach lowers infectious does - Presentation: self-limited acute enterocolitis (6-48hrs post ingestion, lasting 3-7 days), watery stools of moderate volume (occasionally low), tenesmus (painful straining), frequent fever/chills, abdominal cramping, N/V - Tx: abx not recommended in uncomplicated cases. Bacteremia and endovascular invasion may occur in elderly/immunocompromised |
|
|
STEC (including EHEC)
|
Epid: food, water, p2p infection; major cause of bloody diarrhea in developing countries
Clinical: watery/bloody diarrhea, may be complicated by hemorrhagic uremic syndrome (HUS) Pathogenesis: shiga toxins, EHECs also with intimin-Tir-mediated attaching and effacing Prevent: cook ground beef, avoid food cross-contamination, hand washings after farm animal contact, diaper hygiene at pools Tx: supportive care, NO antibiotics (may increase toxin production/HUS) |
|
|
ETEC
|
Epid: infection form contaminated food/water, traveler’s and childhood diarrhea in developing countries
Clinical: acute, watery diarrhea, occasionally severe Pathogenesis: fimbrial adhesins, heat-stable and heat-labile enterotoxins Prevention: bottled water, avoid ice/uncooked foods, bismuth sabsalicylate tablets Tx: hydration, antimotility meds, fluorquinolones |
|
|
E. coli strains
|
General structure: cell envelope, capsule with lipopolysaccharide ) side chains, flagella
ETEC: enterotoxigenic (traveler’s) EPEC: enteropathogenic (childhood dysentery, resource poor areas) EIEC: enteroinvasive (like shigella dysentery) EHEC: enterohemorrhagic (hemorrhagic colitis, hemolytic uremic syndrome), shiga-like toxin production (STEC). Eg: O157:H7 |
|
|
Toxins involved in the pathogenesis of diarrhea
|
Enterotoxins: act on epithelial cells of the small intestine. Produce no structural damage but cause watery diarrhea via increase electrolyte secretion (non-inflammatory). Ex: cholera toxin, ETEC heat-labile toxin (LT)
Cytotoxins: cause damage to the mucosa, resulting in inflammatory colitis. Also can inhibits protein synthesis. Ex: shiga toxin, shiga-like toxin (verotoxins), C. diff cytotoxin Neurotoxins: cause enteric symptoms by targeting the autonomic nervous system. Ex: staph enteroxin B, clostridium botulinum, Bacillus ceres |
|
|
Types of acute infectious diarrhea
|
Non-inflammatory: symptoms are from enterotoxin mediated secretion of eletrolytes/fluid. Typically small bowel (watery diarrhea). Orangisms; cholera, ETEC, EPEC, rotavirus, norovirus, pre-formed toxins
Inflammatory: symptoms are from cytotoxins which stimulate leukocytes (present in stool). The mucosal layer is damaged (usually distal small bowel or colon) causing dysentery. Orgnaisms: shigella, salmonella, campylobacter jejuni, EHEC, Yersinia, vibrio parahaemolyticus, C. diff Penetrating: caused by agents that invade the intestinal mucosa and proliferate in lymphatic or reticuloendothelial cells causing enteric/typhoid fever. Pathogen toxins cause diarrhea by different mechanisms. Organisms: salmonella Typhi, Yersinia enterocolitica |