![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
59 Cards in this Set
- Front
- Back
|
Alagille
|

|
|
|
Alagille syndrome |

Biliary hypoplasia maybe syndromic or non syndromic |
|
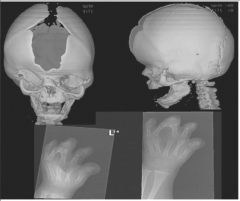
Apert's |

|
|
|

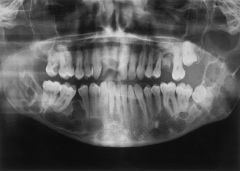
- Autosomal dominant disorder with multiple - keratocystic odontogenic tumors (KOT), basal cell carcinomas (BCCa), and other skeletal and craniofacial abnormalities. - Gorlin goltz/ bifid rib syndrome - Associated abnormalitiesMultiple basal cell carcinomasMarked calcification of falx cerebriRib and vertebral abnormalitiesFrontal and parietal bossing, macrocephaly, hypertelorism. Medulloblastoma. Short fourth |
|
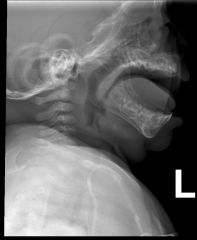
Beckwith-Wiedemann syndrome |

|
|
|
Bechets |

|
|
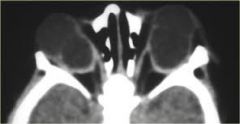
CHARGE
|
Coloboma can be part of the CHARGE syndrome:• Coloboma• Heart anomalies• choanal Atresia• Retardation of growth and development• Genital and Ear anomalies.
|
|
|
COACH
|
COACH syndrome:• Cerebellar vermis hypoplasia,• Oligophrenia (MR)• congenital Ataxia• Coloboma• Hepatic fibrosis.
|
|
|
Crigler–Najjar syndrome
|
- rare - Glucuronosyltansferase. - inherited form of non-hemolytic jaundice- which results in high levels of unconjugated bilirubin - leads to brain damage in infants.- divided into type I and type II, with the latter sometimes called Arias syndrome. - These two types, along with Gilbert's syndrome, Dubin–Johnson syndrome, and Rotor syndrome,
|
|
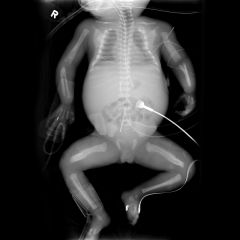
Caffey Disease |
Caffey DiseaseInfantile cortical hyperostosis is a self-limited inflammatory disorder of infants Rare disease manifests at birthcause is unknownPainful periostitis of long bonesInvolvement of clavicle & mandible is suggestive of diagnosis since not usually seen with other diagnosesSelf-limited; spontaneously resolves over first 2 years of life
|
|
|
Caplan syndrome
|
Caplan's syndrome (or Caplan disease or Rheumatoid pneumoconiosis[1]) is a combination of rheumatoid Rarthritis (RA) and pneumoconiosis that manifests as intrapulmonary nodules
|
|
|
CCD |

|
|
|
Carney complex
|

|
|
|
Coats disease |

|
|
|
Connective tissue disorders |
Marfan Syndrome • Rare genetic disorder secondary to fibrillin-1 gene mutation • Systemic abnormalities: Most commonly aortic aneurysm/dissection, scoliosis, pectus excavatum, dural ectasia, ectopia lentis, tall slender buildEhlers-Danlos Syndrome • Rare genetic disease resulting in skin hyperextensibility, easy bruising, joint hypermobility • Mutation of collagen V genesCutis Laxa • Rare genetic disease resulting in wrinkled & redundant skin, emphysema, hernias, diverticula of the GI & GU tracts • Mutation to the elastin genesBirt-Hogg-Dubé Syndrome • Rare autosomal dominant disease: Renal cell carcinoma, cutaneous fibrofolliculoma, pulmonary cysts • Mutation of folliculin gene |
|
|
Cronkhite-Canada |
- Generalized GIT polyps |
|
|
Crouzon's syndrome
|
-complex syndromic synostosis-maxillary hypoplasia-Limbs are usually clinically normal
|
|
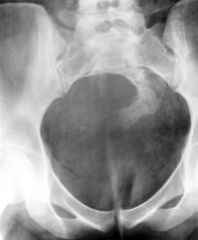
Currarino's triade |
Anorectal malformation (commonly anorectal stenosis) Bony sacral anomalies (classically a ‘scimitar sacrum’ with unilateral hypoplasia ofthe lateral aspect of the vertebral bodies) Presacral mass lesion (amt meningocele or teratoma)
|
|
|
Di George syndrome |
- 22q11 deletion syndrome- 3rd and 4th Pouch AnomaliesCATCH-22Cardiac Abnormality (tetralogy of FallotAbnormal faciesThymic aplasiaCleft palateHypocalcaemia /Hypoparathyroidism.
|
|
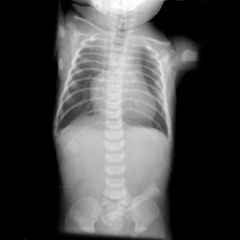
Down
|
MSK• Atlanto-axial subluxation and instability with hypoplasia of the odontoid• vertebral bodies are relatively tall• 11 ribsand the ribs themselves are gracile• multiple manubrial ossification• iliac wings are flared with relatively sloping• hands are short with clinodactyly of the little finger (hypoplastic middle phalanx)• bilateral clubbed feetCVS• ASD(ostium primum)• Congenital heart lesions include • endocardial cushion defects GIT• TOF• Duodenal atresia, duodenal stenosis, • Hirschsprung's disease• anorectal anomalies are associated.CNS• Wormian bones. - Moyamoya
|
|
|
Dwarfism |
locationrhizomelic = root, e.g., bones of the upper arm or thighmesomelic = middle, e.g., bones of the forearm or lower legacromelic = end, e.g., bones of hands and feet.micromelic = entire limbs are shortenedsourcechondro = of cartilageosteo = of bonespondylo = of the vertebraeplasia = formtrophy = growth
|
|
|
ellis van creveld
|
ARalso called chondroectodermal dysplasia or mesoectodermal dysplasiashort limbtibial hypoplasiashort limb not trunk involvmentpolydactly of handcommon pathologies of ellis van creveld syndromecongenital heart disease (common atrial chamber)septal defectsnail dysplasia at birth
|
|
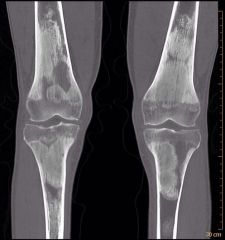
ERDHEIM-‐CHESTERDISEASE |
Lipidgranulomatosiswithretroorbitaldeposition,xanthelasmaofeyelids,skeletalmanifestations(medullarysclerosis,corticalthickening),andcardiopulmonarymanifestationsduetocholesterolemboli.
|
|
|
Edwards syndrome / Trisomy 18 |
kidney malformations, structural heart defects at birth (i.e., ventricular septal defect, atrial septal defect, patent ductus arteriosus), intestines protruding outside the body (omphalocele), esophageal atresia, intellectual disability, developmental delays, growth deficiency, feeding difficulties, breathing difficulties, and arthrogryposis- club foot- rocker bottom. - horse shoe kid
|
|
|
Fanconi anaemia |
FA is the result of a genetic defect in a cluster of proteins responsible for DNA repair. As a result, the majority of FA patients develop cancer, most often acute myelogenous leukemia, and 90% develop bone marrow failure (the inability to produce blood cells) by age 40. About 60–75% of FA patients have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities.
|
|
|
Fanconi syndrome
|
Fanconi syndrome (also known as Fanconi's syndrome) is a disease of the proximal renal tubules[1] of the kidney in which glucose, amino acids, uric acid, phosphate and bicarbonate are passed into the urine, instead of being reabsorbed. Fanconi syndrome affects the proximal tubule, which is the first part of the tubule to process fluid after it is filtered through the glomerulus. It may be inherited, or caused by drugs or heavy metals.[2]
|
|
|
Gilbert syndrome
|
- most common hereditary cause of increased bilirubin - up to 5% of the population - unconjugated hyperbilirubinemia- reduced activity of the enzyme glucuronyltransferase, which conjugates bilirubin - Conjugation renders the bilirubin water-soluble, after which it is excreted in bile into the duodenum.
|
|
|
Gardner's syndrome
|
o FAPo Extracolonic manifestations are really just part of thespectrum of FAP Multiple osteomas of the skull and mandible Epidermoid cysts Soft-tissue tumours Abnormal dentition Desmoid tumour formation- medulloblastomas- Adrenal, thyroid, ampullary, and liver carcinomas- Congenital pigmented lesions of retina
|
|
|
Holt–Oram syndrome |
absent radial bone in the armsatrial septal defectfirst degree heart blockconsidered to be a phenocopy of thalidomide. -A1 syndrome
|
|
|
Hajdu–Cheney
|
acroosteolysis with osteoporosis and changes in skull and mandible extremely rare autosomal dominant 70 cases have been reported worldwide.
|
|
|
Henoch–Schönleinpurpura(HSP) |
skinrash,abdominalpain,arthralgia,andnephritis
|
|
|
Jaffe-Campanacci syndrome
|
Presentations may include mental retardation, disseminated non-ossifying fibromas of the long bones and jaw, hypogonadism or cryptorchidism, or giant cell granulomas of the jaw.[1]
|
|
|
Klippel-Feil Spectrum
|
-Congenital spinal malformation characterized by segmentation failure of 2 or more cervical vertebrae ± thoracic, lumbar segmentation failureAssociated abnormalities- Hemivertebrae, butterfly vertebrae, spina bifida- Scoliosis (usually congenital) ± kyphosis (60%)- Odontoid dysplasia, basilar impression, C1 assimilation, occipito-cervical instability- Syringomyelia, diastematomyelia (20%), Chiari 1 malformation (8%), neurenteric cyst or dermoid (rare)- Cervicomedullary neuroschisis ± synkinesis (20%)- Sprengel deformity ± omovertebral bone (15-30%); unilateral or bilateral- Sensorineural hearing loss (30%), external ear anomalies, - GU tract abnormalities (35%), - congenital heart disease (14%), - upper extremity deformity, facial anomalies
|
|
|
Lesch–Nyhan syndrome
|
also known as juvenile goutdeficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRTmutations in the HPRT gene located on (the) X chromosome. self mutilation
|
|
|
Leriche syndrome
|
: Abdominal aortic or iliac artery occlusive disease causing absent femoral pulses, bilateral claudication and impotence
|
|
|
Lemierre's syndrome
|
- Lemierre syndrome refers to rare thrombophlebitis of the jugular veins with distant metastatic sepsis seen in the setting of initial oropharyngeal infection (pharyngitis / tonsillitis +/- peri tonsillar abscess). - Fusobacterium necrophorum
|
|
|
Lutembacher syndrome
|
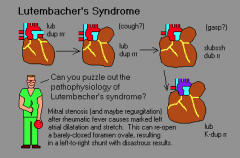
|
|
|
Marfan's
|
Autosomal dominant connective tissue disease, Fibrillin gene defect on chromosome 15 VASC 60%: - Ascending aorta aneurysm - Aortic regurgitation/root ectasia (common) - Dissection - Mitral valve prolapse and regurgitation- Pulmonary artery aneurysmMSK- Scoliosis- Pectus excavatum- Arachnodactyly- Acetatabular protrusion- Pes planusSPINE- Dural ectasia- Anterior meningocoele- Arachnoid cysts- Scoliosis- Vertebral scalloping- Enlargement of sacral foraminaCHEST- Superior rib notching- Recurrent pneumothora- interstitial lung disease or bronchiectasisEYE-lens dislocation- High palate
|
|
|
MEN
|
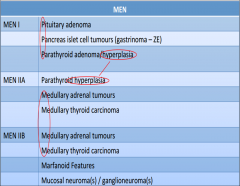
|
|
|
Morquio |

Madelung more Hurler |
|
|
Niemann–Pick disease
|
- Lipid storage disorder 2° to deficiency of lysosomal sphingomyelinase (lysosomal storage diseases)- hepatosplenomegaly- Basal ganglia- Marrow packing (similar radiographic features as gauchers)
|
|
|
Noonan Syndrome
|
congenital heart defect (typically pulmonary valve stenosis) also ASD, hypertrophic cardiomyopathy, short stature, learning problems, pectus excavatum, impaired blood clotting, and a characteristic configuration of facial features including a webbed neck and a flat nose bridge.
|
|
|
OI |

|
|
|
OWR/Hereditary hemorrhagic telangiectasia
|
Hereditary epistaxismuco cutaneous telangiectasisMultiple AVM, lung liver, brain, kidney, GIT,pancreas
|
|
|
OEIS
|
(omphalocele, exstrophy of the cloaca, imperforate anus, spinal defects)
|
|
|
POEMS
|
Polyneuropathy Organomegaly Endocrinopathy Monoclonal gammopathy/multiple myeloma Skin changes
|
|
|
Pompe
|
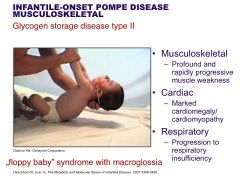
|
|
|
Prune Belly
|
Characterized by 3 principle componentsDramatic collecting system dilatation(Urethral dysplasia)Deficiency of abdominal musculatureCryptorchidism
|
|
|
Potter sequence
|
- Physical appearance of a fetus or neonate due to oligohydramnios experienced in the womb- Oligohydramnios is the causative agent of Potter sequence, but there are many things that can lead to oligohydramnios.
|
|
|
Psoriasis 5 types
|
classic/polyarthritis(acroosteolysis). arthritis mutilanssymmetric polyartritis(RA variant)oligoarticular(most common/asymmetric)spondyloarthropathy
|
|
|
SAPHO
|
- synovitis, acne, pustulosis, hyperostosis, osteitis- CRMO is a varient in children, with blisters on their hands and feet
|
|
|
Shone syndrome
|
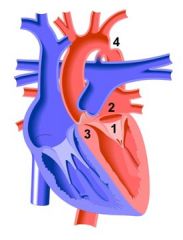
Shone syndrome is characterised by four congenital heart defects, largely multiple left sided obstructions: supravalvular mitral membrane (SVMM) subaortic stenosis (membranous or muscular)parachute mitral valvecoarctation of the aorta
|
|
|
Turcott
|
-Mismatch repair cancer syndrome (MMRCS) - Brain tumor-polyposis syndrome- Glioma-polyposis syndrome- medulloblastoma, malignant glioma.
|
|
|
Trevor-Fairbank disease
|
Dysplasia Epiphysealis Hemimelica-developmental disorder characterized by asymmetric cartilaginous overgrowthof one or more epiphyses in the lower extremity-Pathologically, similarity to an osteochondroma, and for this reason it isoccasionally referred to as “epiphyseal” or “intraarticular osteochondroma.”
|
|
|
Turner XO
|
45-XO karyotypeMSK• Short stature and lymphoedema may be clinically obvious.• Short fourth metacarpals, • Madelung deformity• Flattening of the medial tibial condyle with a transitory exostosis, • Osteoporosis, • Scoliosis, • Delay in maturation of the skeleton.CVS• Coarctation of the aorta, GU• Urinary tract anomalies, such as horseshoe kidneys (15%)• Small uterus• Classical form of ovarian dysgenesis• Elevated follicle-stimulating hormone (FSH) concentrations• ‘Streak ovaries’GENRAL• Widely spaced nipples• Poorly developed breasts• High arched palate• Webbed neck• Low posterior hairline• Pigmented nevi
|
|
|
Treacher Collins |
|
|
|
VHL
|

|
|
|
Williams syndrome |
- neurodevelopmental disorder- craniofacial dysmorphism (e.g. elfin facies)- supravalvular aortic stenosis/ pulmonary- transient hypercalcaemia
|
|
|
WAGR syndrome |
Wilms tumor, aniridia, genitourinary anomalies (cript most common), and mental retardation syndrome
|