![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
178 Cards in this Set
- Front
- Back
|
Diagramthe structure of a mitochondrion, locating the major features including thematrix, the inner and outer membranes, the cristae, and the site of the TCAcycle enzymes. |
1. Matrix: all TCA cycle rxns occur within -within IM 2. OM: permeable to many cmpds b/c has pores 3. IM: impermeable to everything (including H+, some H2O and gases may slip through) → specific carriers transport molecules across it -foldings = cristae *source of E for the cell via oxidative phosphorylation |
|
|
Describe the steps in the TCA cycle that yield CO2,NADH, FADH2, and GTP. |
***CO2: 1. isocitrate dehydrogenase 2. α -ketoglutarate dehydrogenase (PDH) NADH: 1. isocitrate dehydrogenase 2. α -KGDH 3. malateDH (PDH) FADH2: succinate DH GTP: succinyl-CoA synthetase |
|
|
Describethe intermediates of the TCA cycle that are precursors for other biosyntheticpathways. |
* |
|
|
Illustratewhere in the TCA cycle that fats, sugars, and amino acids enter the pathway. |
Carbs: enter as Acetyl-CoA FA: oxidation → Acetyl-CoA aa degradation: → TCA intermediates for gluconeogenesis or E production: 1. α -ketoglutarate 2. succinyl-CoA 3. fumarate 4. OAA |
|
|
Describe the regulatory mechanismsthat control the activity of the TCA cycle. How does this regulation coordinatewith the pathways of glycolysis and gluconeogenesis, already described? |
* |
|
|
Explain the need for anaplerotic reactions, and describe themajor anaplerotic reactions. |
* |
|
|
Explain why is it not possible to obtain the net synthesis of glucose from acetyl-CoA,despite the fact that labeled carbon atoms from acetyl-CoA can be incorporatedinto glucose? Explain how such carbonatoms can end up in glucose. |
* |
|
|
Describethe vitamins that are required for the pyruvate and a-ketoglutaratedehydrogenase reactions. Which diseasesmay result from a deficiency of these vitamins? |
* |
|
|
Explain the concept of ΔG0and ΔG0’. How does one use the information from ΔG0 todetermine in which direction a reaction will proceed? How does the concentration of the reactantsfactor into this determination? Explainhow standard conditions are different from cellular conditions. |
* |
|
|
Explainhow mutations in isozymes of TCA cycle enzymes have been linked to cancer. |
* |
|
|
Role of TCA Cycle |
Central point of metabolism: 1. Generate E by oxidizing acetyl-CoA to CO2 and H2O -- e- must be removed from the 2 Cs → accepted by NADH (6) and FADH2 (2) --acetyl-CoA is produced from pyruvate, FA ox, aa ox 2. Supply biochemical intermediates for: a. heme synthesis b. aa synthesis 3. Entry point for carbs, FAs, aas |
|
|
Patient has genetic defect that causes intestinal epithelial cells to produce disaccharidases of much lower activity. After eating bowl of sugary cereal with milk, patient will have higher levels of which of the following? |
maltose, sucrose, lactose in the stool |
|
|
young infant nourished by synthetic formula had sugar in blood and urine that gave positive reducing sugar test but didn't show positive for glucose test. Which cmpd? |
Fructose (reducing sugars have a carbonyl) |
|
|
What tissue will be responsible for hypoglycemia experienced when a patient injects insulin but doesn't eat a meal after. |
Adipose/muscle |
|
|
How many e- will be captured to full oxidize carbons to in Acetyl-Coa to CO2 and H2O? |
8 e- (2Cs x 4e-) |
|
|
What determines if a rxn is favorable? |
Thermodynamics amount of E required/released by a rxn → Gibbs Free E |
|
|
Gibbs Free Energy |
Δ G= Δ H - T Δ S Δ H = change in enthalpy Δ S = change in entropy Δ G = (-) → rxn proceeds in forward direction |
|
|
Δ G^o |
Free E change of a rxn under standard conditions: 298 K = 25 oC 1 atm P all reactants and products at initial conc = 1.0 M |
|
|
Δ G ^o' |
Biochemical Standard of Gibbs Free E Free E chnage at: pH = 7.0 H2O= 55.5 M all other reactants and products at initial conc. = 1.0M (result of water not being able to be 1.0 M and the effect pH has on bio rxns) |
|
|
Δ G = |
A+B → C+D Δ G =Δ G^o' + RTln [C][D]/[A][B] factors for when aren't under standard conds. R= 1.987 cal/mol-K (gas constant) T=kelvin Δ G (-) → rxn proceeds (look up Δ G^o' in a table) |
|
|
Coupled rxns Δ G^o' |
can be added together |
|
|
kJ kcal conversion |
4.18 kJ = 1 kcal |
|
|
how (+) Δ G^o' rxn proceeds |
small product/reactant ratio → (-) RTln |
|
|
pyruvate transport into the mito |
(is created in cytoplasm→ must be transported in) -crosses through pores of OM -two transporters in IM: 1. Symport: (2o active transport) -protons simultaneously pumped in down their conc grad w/ pyruvate 2. Antiport: -citrate (3 neg. charges) out, pyruvate (1 - charge) in -also driven by proton gradient (electrical grad) |
|
|
fates of pyruvate |
1. lactate 2. alanine in mito: 3. OAA 4. Acetyl-CoA → can no longer be used to make glucose *why FAs can't be used to make glucose |
|
|
Pyruvate dehydrogenase |
Pyruvate + CoA + NAD+ → Acetyl-CoA + CO2 + NADH + H+ -oxidative decarboxylation= substrate is α -ketoacid -enzyme has five subunits* |
|
|
Subunits of Pyruvate Dehydrogenase |
2 regulatory 3 catalytic: 1. pyruvate decarboxylase (E1): binds thiamin pyrophasphate (B1) 2. Transacetylase (E2): utilizes lipoic acid 3. Dihydrolipoyl dehydrogenase (E3): has bound FAD, **shared w/ other enzymes that perform oxidation decarboxylation rxns (E3 mutation affects all of these) |
|
|
Cofactors required by PDH |
1. Thiamine (B1) → Thiamin pyrophosphate 2. lipoic acid 3. FAD 4. coenzyme A |
|
|
Thiamin pyrophosphate |
-derived from thiamine (B1) via phosphorylation (ATP → AMP) adding two P groups on - Thiazolium ring has acidic H that is easily lost → helps break C-C bonds of two structure types: 1. α - ketoacids (bw/n two carbonyls) 2. carbohydrates (bw/n carbonyl C and α C connected to hydroxy) |
|
|
FAD |
flavin - D- Ribitol - pyrophosphate - D-Ribose - Adenine (flavin-ribitol-ADP) -ribitol (reduced ribose) and flavin derived from riboflavin (B2) -stronger oxidizing agent than NAD+ → FADH2 is a weaker reducing agent than NADH |
|
|
FAD e- acceptance |
-1 e- + H+ at a time (vs. hydride transfer = two at a time in NAD) |
|
|
Coenzyme A (CoA) |
-derived from pantothenic acid (phosphopantothene) -provides active sulfhydryl to conserve E in a thioester linkage in pyruvate dehydrogenase rxn |
|
|
Lipoic Acid |
-five-membered ring w/ disulfide bond -pyruvate dehydrogenase rxn: --disulfide reduced then oxidized |
|
|
Arsenite (Arsenic) |
interferes w/ cmpds containing adjacent sulfhydryl groups : -lipoic acid → pyruvate dehydrogenase, α -ketoglutarate DH |
|
|
why pyruvate dehydrogenase is so highly regulated |
once acetyl-CoA is formed, it cannot be used to produce glucose |
|
|
rxns that share E3 subunit (Dihydrolipoyl dehydrogenase) |
catalyze oxidation decarboxylations 1. TCA: -a. pyruvate dehydrogenase -b. Isocitrate dehydrogenase????? -c. α -ketoglutarate dehydrogenase 2. branched-chain aa metabolism 3. α - ketobutyrate → propionyl-CoA *mutations in this subunit affect all of these rxns |
|
|
Pyruvate Dehydrogenase Complex deficiency |
1. lactic acidemia 2. neuro probs (brain dependent on glucose) -X-linked dom: E1 α subunit located on X-chrom → most common defect |
|
|
Thiamine Deficiency Disorders |
1. Beriberi- alcoholism, malabsorption (alcohol blocks thiamin (B1) uptake from gut) *individuals usually get one or the other, don't know what determines which a. Dry b. Wet 2. Wernicke-Korsakoff Syndrome- brain disorder, lack of thiamin a. Wernicke encephalopathy b. Korsakoff syndrome |
|
|
Dry Beriberi |
affects NS: 1. difficulty walking 2. loss of feeling in hands and feet 3. mental confusion/speech difficulties 4. Nystagmus 5. tingling 6. vomiting (7. coma 8. death) |
|
|
Wet Beriberi |
cardiovascular system: 1. awakening at night short of breath 2. increased heart rate 3. shortness of breath w/ activity 4. swelling of lower legs 5. can lead to congestive heart failure |
|
|
Wernicke encephalopathy |
-result of beriberi going on for too long -symptoms may be reversed if given thiamine 1. confusion, loss of mental activity (→ coma, death) 2. loss of mm. coordination, leg tremors 3. Nystagmus, double vision, eyelid droop 4. alcohol w/drawal symptoms |
|
|
Korsakoff syndrome |
-evolves from Wernicke encephalopathy -not usually reversible 1. inability to form new memories 2. loss of memory 3. confabulation (making up stories) 4. hallucinations |
|
|
Which vitamin is not needed for TCA cycle? |
vitamin C |
|
|
TCA cycle enzymes |
1. Citrate Synthase 2. Aconitase 3. Isocitrate dehydrogenase 4. α -ketoglutarate dehydrogenase 5. Succinyl-CoA synthetase 6. Succinate Dehydrogenase 7. Fumarase 8. Malate dehydrogenase |
|
|
Citrate synthase |
Acetyl-CoA + Enz-B: → nuc-Acetyl-CoA nuc-Acetyl-CoA + OAA → Citroyl-CoA → Citrate + HSCoA -Citroyl-CoA: --not found in soln, only bound to enzyme --high E thioester → hydrolysis makes rxn favorable Δ G = -8 kcal/mol, Not reversible→ OAA levels stay low |
|
|
Aconitase |
citrate ↔ (H2O out) + Cis-Aconitate (H2O in) ↔ Isocitrate Citrate: -symmetrical -aconitase treats dehydrates and rehydrates asymmetrically→ always the same bond Δ G^o' = (+) ONLY TCA rxn positive under cellular conds 90:6:4 ratio, rapid utilization of isocitrate allows rxn to proceed |
|
|
Isocitrate dehydrogenase |
isocitrate + Enz-B: + NAD+ → NADH + oxalosuccinate → CO2 + α -ketoglutarate -oxidation-decarboxylation -very different rxn mechanism from pyruvate dehydrogenase -use E3?? Δ G^o' = -5 kcal/mole → helps drive aconitase rxn |
|
|
α -ketoglutarate dehydrogenase |
α -KG + NAD+ + CoA → Succinyl-CoA + NADH + CO2 -oxidative decarboxylation -uses E3 (same cofactors as PDH!) Δ G^o' = -8 kcal/mole |
|
|
Succinyl-CoA Synthetase (succinate thiokinase) |
Succinyl-CoA + Pi + GDP ↔ succinate + GTP + CoA -Pi displaces CoA → high E bond → substrate level phosphorylation -Δ G^o' = -0.8 kcal/mole → easily reversible |
|
|
nucleoside diphosphate kinase |
GTP + ADP ↔ GDP + ATP |
|
|
Overall rxn and net yield of TCA up to Succinyl-CoA Synthetase |
AcCoA + OAA + 2NAD+ + GDP + Pi + CoA → succinate + 2CO2 + 2NADH + GTP + 2CoA -2 carbons have been lost -4 e- have been lost - 1 GTP (→ ATP) has been gained |
|
|
Succinate Dehydrogenase |
succinate + FAD ↔ fumarate + FADH2 -oxidation of C-C bond → requires strong oxidizing agent = FAD **enzyme is attached and embedded within inner mito membrane=not found in matrix |
|
|
Fumarase |
fumarate + H2O ↔ Malate (hydroxyl group replaces c=c bond in fumarate) |
|
|
Malate dehydrogenase |
malate + NAD+ ↔ OAA + NADH Δ G^o' = 29.7 kJ/mol -cellular conds: low intramito [OAA] from high activity of citrate synthase and use of OAA for gluconeogenesis |
|
|
TCA cycle rxn that under cellular conditions has (+) Δ G |
aconitase citrate ↔ (H2O out) + Cis-Aconitate (H2O in) ↔ Isocitrate |
|
|
TCA cycle rxns that have (+) Δ G^o' |
aconitase (+6.3 kJ/mole) Malate dehydrogenase (+29.7 kJ/mole) |
|
|
net Δ G ^o' of TCA |
-57.3 kJ/mole |
|
|
Overall TCA cycle rxn |
Acetyl CoA + 2H2O +3NAD+ + FAD + GDP + Pi → 2CO2 + 3NADH + 3H+ + FADH2 + CoA + GTP |
|
|
E generation in per 1 mole of acetyl-CoA oxidized in TCA cycle |
10 moles high E bonds: (9 ATP, 1 GTP) 3 NADH x 2.5 ATP = 7.5 1 FADH2 x 1.5 ATP = 1.5 1 GTP |
|
|
What i the yield of ATP when 1 molecule of fructose is ocmpletely oxidized to CO2 and H2O? |
32 ATP (OR 30) same for glu and gal |
|
|
Overall equation and yield of total oxigation of glucose |

(or 30 ATP if less efficient NADH transporter used) |
|
|
yield of complete oxidative of glucose compared to glyolysis |
16 x BUT glycolysis much faster → mm. use glycolysis when exercising |
|
|
Major regulatory step of TCA cycle |
PDH (which isn't actually in cycle) |
|
|
regulation of pyruvate dehydrogenase |
Inhibitors: Allosteric: (via products) 1. acetyl-coA (competive inhibitor of CoA) 2. NADH (competive inhibitor of NAD+) Covalent: 3. phosphorylation by PDH kinase → PDH-b Activators: 1. PDH phosphatase → PDH-a 2. (CoA, NAD+, Ca2+) |
|
|
PDH kinase |
PDH-a + ATP → PDH-b + ADP -phosphorylates pyruvate decarboxylase (E1) subunit at 3 Ser -distinct from cAMP dependent protein kinase (location= mito vs. cyto) Activators: (same as allosteric inhibitors of PDH) 1. NADH 2. Acetyl-CoA Inhibitors: 1. Pyruvate 2. ADP |
|
|
PDH-a vs. PDH-b |
PDH-a: active, not phsophorylated PDH-b: inactive, phosphorylated |
|
|
PDH phosphatase |
PDH-b + H2O → PDH-a + Pi Activators: 1. Ca2+ 2. Insulin |
|
|
What other enzymatic activity is inhibited by direct phosphorylation? |
liver pyruvate kinase |
|
|
TCA cycle regulation |
1. Pyruvate DH** 2. Citrate Synthase 3. Isocitrate dehydrogenase 4. α -ketoglutarate dehydrogenase 8. Malate dehydrogenase *NADH is key player → enzymes that produce NADH will be inhibited by it |
|
|
Citrate synthase regulation |
inhibited by citrate |
|
|
isocitrate dehydrogenase regulation |
Activators: ADP Ca2+ Inhibitors: NADH **regulated so that citrate can accummulate b/c aconitase favors citrate formation→ signifies adequate building blocks → transported to cyto and inhibits PFK-1 (slowing glycolysis) and activates FA synthesis (citrate is substrate for FA production) |
|
|
α -ketoglutarate dehydrogenase regulation |
Activators: Ca2+ Inhibitors: NADH **regulated b/c can form glutamate in one step → leaves cycle; encouraged if excess E is present |
|
|
malate dehydrogenase regulation |
inhibited by NADH |
|
|
Malonate |
disrupts TCA cycle: competitive inhibitor of succinate DH (similar structure to succinate minus 1 methylene) |
|
|
Arsenite |
disrupts TCA cycle: disrupts enzymes requiring lipoic acid: 1. PDH 2. α-ketoglutarate DH |
|
|
Fluoroacetate |
disrupts TCA cycle: produces fluorocitrate → blocks aconitase |
|
|
Anaerobosis (lack of oxygen) |
disrupts TCA cycle: NADH accummulates in mito → NADH producing enzymes inhibited |
|
|
Inhibitors of e- chain effect on TCA cycle |
disrupts TCA cycle: NADH accumulates → NADH producing enzymes inhibited |
|
|
Uses for Acetyl-CoA |
1. cholesterol synthesis 2. FA synthesis 3. oxidation for E CANNOT be used as a precursor to gluconeogenesis |
|
|
Why acetyl-CoA can't produce net glucose |
-requires it to become pyruvate or OAA -PDH is irreversible -is 2 Cs → condenses w/ OAA → 2Cs lost as CO2 → no net synthesis of OAA only its regeneration -no mammalian pathwyas for production of OAA from 2 acetyl-CoA b/c we are missing two enzymes necessary --glycoxylate cycle in plants and bacteria can do this |
|
|
Rate limiting factor of TCA cycle |
OAA: OAA + AcCoA → citrate (very favorable) OAA + NADH → malate + NAD+ (favorable direction) these two rns redice OAA levels w/n TCA cycle → OAA stays at low levels → keeps malate dehydrogenase favorable under cellular conds → Anaplerotic rxns replenish OAA so cycle can continue |
|
|
Loss of TCA cycle intermediates |
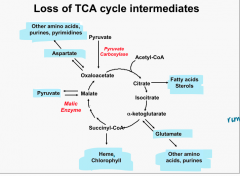
loss of intermediates → cycle slowed |
|
|
TCS cycle Anaplerotic Rxns |
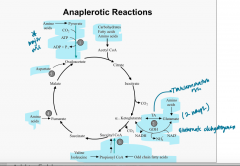
|
|
|
Loss of citrate |
citrate → fatty acids, sterols |
|
|
Loss of α -ketoglutarate |
α -ketoglutarate ↔ glutamate → other aas, purines |
|
|
Loss of succinyl-CoA |
succinyl-CoA → Heme, Chlorophyll |
|
|
Loss of Malate |
Malic Enzyme: malate ↔ pyruvate |
|
|
Loss of OAA |
OAA ↔ Aspartate → other aas, purines, pyrimidines |
|
|
Anaplerotic rxns |
-refilling rxns for TCA cycle 1. OAA*** (major one via pyruvate) 2. Acetyl-CoA 3. α -ketoglutarate 4. succinyl CoA 5. Fumarate |
|
|
Anaplerotic rxns: OAA |
glycolyisis → pyruvate → OAA via pyruvate carboxylase Transaminations from aas: alanine → pyruvate → OAA (via pyruvate carboxylase) aspartate → OAA |
|
|
Anaplerotic rxns: Acetyl-CoA |
Carbs, FAs, aas → Acetyl-CoA |
|
|
Anaplerotic rxns: α -ketoglutarate |
aas → glutamate → α -ketoglutarate via: 1. transamination rxn 2. glutamate dehydrogenase (NAD+ in, NADH and NH4+ out) |
|
|
Anaplerotic rxns: Succinyl-CoA |
Valine, Isoleucine → propionyl CoA → Succinyl-CoA Odd chain FAs→ propionyl CoA → Succinyl-CoA |
|
|
Anaplerotic rxns: Fumarate |
aas → fumarate |
|
|
Regulation of TCA cycle tied to Glycolysis |
high E levels (increased NADH) → isocitrate DH inhibited → citrate accumulation → citrate leaves mito → PFK-1 inhibited → E storage begins (fat and glycogen synthesis) |
|
|
PDH and pyruvate decarboxylase regulation |
reciprocal: high [Acetyl-CoA]: PDH inhbited, pyruvate decarboxylase activated → more OAA produced → 1. condenses w/ excess acetyl-CoA for FA synthesis (high BG) OR 2. used for gluconeogenesis (low BG) (depends on cell's needs) |
|
|
which TCA cycle intermediate will accummulate in the presence of arsenite? |
α -ketoglutarate |
|
|
Arsenate will inhibit which TCA rxn? |
succinyl-CoA synthetase (inhibits free Pi rxns) |
|
|
Pharmacological treatment can help with deficieny of which enzyme? |
citrate synthase???????????*********** |
|
|
role of oxidative phosphorylation |
-generate E as ATP from e- flow: --e- flow from reducing agent (NADH, FADH2) → more stable carrier (O2 → H2O) --as e- flow to most stable form E is released and captured as a proton gradient --proton flow down electrochemical gradient drives ATP synthesis (E captured w/n high E bonds of ATP) = hemiosmotic hypothesis (theory) |
|
|
Proton gradient |
e- flow pumps H+ from mito matrix across IM to intermembrane space |
|
|
Calculating E yield for proton gradient |
Nernst: ΔGo = -(n)(F)(Δ Eo') n= # of e- transferred F= Faraday constant = 23 kcal/mol-volt Δ Eo'= change in redox potential under standard conditions (all reactants at 1.0 M concentrations) |
|
|
A drug results in elevated lactate and α-ketoglutarate. Drug inhibits rxns which require which vitamin? |
Pantothenate? (PDH AND α -ketoglutarate inhibited=can't be biotin) |
|
|
Conversion of fructose to pyruvate under aerobic conds results in? |
2 ATP, 2 NADH |
|
|
What type of gradient is generated by protons? |
electrical gradient and pH gradient (and chemical?) |
|
|
Redox potential (Δ E) |
measures substrate's affinity for e- as compared to standard rxn : 2H+ + 2e- → H2 Eo'= 0 volts (-)Eo': e- donor stronger than H2=e- flow from NADH to H+ (+) Eo': e- donor weaker than H2 = will accept e- from H2 |
|
|
E released when NADH donates e- to O2: calculating Δ Eo' |

|
|
|
E released when NADH donates e- to O2: calculating Δ G |

|
|
|
e- transport chain efficiency |
synthesis of 1 high E bond of ATP requires 12kcal/mol under physion conds e- transfer from 1NADH = -52.3 kcal/mol → potential to make 4 high E bonds per e- pair BUT only make 2.5 ATP = 60% efficiency other 40% lost as heat |
|
|
Eo' and e- flow |

flows form low to high (negative to positive) |
|
|
Given these redox potentials: A +0.45 B - 0.13 C + 0.77 D -0.32 order of e- flow? |
D→B→A→C |
|
|
TCA rxns involving water |
aconitase: dehyrdration, hydration fumarase: hydration |
|

|

|
|
|
Which enzyme will syntthesize a high E bond only in the presence of O2? |
succinate thiokinase (no O2 → NADH built up → TCA cycle stopped) |
|
|
Electron Transport Chain |
1. Complex I |
|
|
Complex I |
NADH:CoQ oxidoreductase/NADH dehydrogenase -NADH converted back to NAD+ → transfers 2 e- to FMN (1 at a time) → e- transfered to Fe-S complexes (6-7) which have different redox potentials (goes from more - to more +) → 1 or 2 e- transferred to CoQ → QH2 -4 H+ pumped from matrix to intermembrane space per 2 e- |
|
|
FMN |
flavin mononucleotide -derived from FAD -flavin + ribitol + P -flavin accepts e- one at a time → radical intermediate formed after 1 e- accepted |
|
|
Fe-S |
iron reduced/oxidized: Fe 3+ ↔ Fe2+ |
|
|
CoQ |
Coenzyme Q = ubiquinol -derived from CoA -can accept 1 or 2 e- → stable as either: QH (radical) or QH2 (completely reduced) |
|
|
What other enzyme requires FAD? |
Pyruvate dehydrogenase Succinate dehydrogenase |
|
|
Complex III |
Ubiquinol-cytochrome C oxidoreductase -QH2 donates 1 e- to CytbL → 2H+ pumped out + QH radical, donates other e- to Cyt C1→ 2H+ pumped out + CoQ formed -Cyt C1 donates e- to Cyt C -CytbL donates e- to CytbH which donates to CoQ and 1 H+ pumped in→ QH radical formed, receives e- from complex I and H+ pumped in → QH2 (total of four H+ pumped out) |
|
|
Problems MI/heart attack causes in e- transport chain |
blood flow greatly decreased → no O2 → everything reduced in chain = QH or QH2 -when O2 is restored QH radical can react w/ O2 → radical oxygen form |
|
|
Complex IV |
Cyt C |
|
|
Cytochromes |
contain heme groups → Fe 3+ + e-↔ Fe 2+ |
|
|
Complex IV |
cytochrome oxidase Cyt C donates e- 1 at a time to Cyta1:CuA → donates e- to Cyta3:CuB → 2 H+ pumped out once 4 e- collected w/n complex IV O2 reduced to H2O: (1/2 O2 + 2H+ → H2O) x2 (b/c 1/2 O2 doesn't actually exist) |
|
|
Cua and Cub |
Cu+2 + e- ↔ Cu +1 |
|
|
Complex II |
contains succinate dehydrogenase -entry point for FADH2 from TCA cycle (weaker reducing agent, enters chain later NADH) -2 e- donated to FAD → Fe-S → CoQ (1 or 2 e- at a time) *no proton extruxion (E transfers are too low) |
|
|
protons pumped from mito matrix to intermembrane space from 1 NADH (2 e-) |
10 H+ |
|
|
protons pumped from mito matrix to intermembrane space from 1 FADH2 (2 e-) |
6 H+ |
|
|
How E us ibtained from the e- transfer chain |
proton gradient established across inner mito membrane: -high [H+] outside than inside -electrical and pH (concentration) gradient - entry of H+ into mito is energetically favorable → 4 H+ required to make 1 high E bond |
|
|
Proton Motive Force |
two components: 1. Δ pH: (pH matrix > pH intermembrane space) 2. Δ ψ: [H+] matrix < intermembrane space) → electrochemical gradient |
|
|
calculating PMF |

|
|
|
ATP synthase |
F0: pore -membrane ebedded -C subunits F1: Headpiece - gamma (assymetric( -α x3 -ß x3 H+ flows through pore down its electrochemical grad → c subunit changes → gamma rotates → α -ß pairs change conformation based on which part of gamma is interacting with it → bound ATP is released, ADP + Pi is converted to bound ATP, new exposed dimer binds ADP (4 H+ = movement of α -ß to next conformation?) |
|
|
which part of ATP synthase requires E? |
Making ATP is reversible release of ATP requires E |
|
|
Oligomycin |
-blocks ATP synthesis by blocking F0 pore (proton channel) → H+ cant go through pore → respiration blocked due to coupling of ATP synthesis and e- flow |
|
|
Principles of Chemiosmotic Hypothesis |
!. e- transfer chain must be assymetrically oriented across the membrane such that protons are ejected 2. Mito must be capable of ejecting protons in the presence of substrate and O2 (measurable decrease in pH outside) 3. permeabilizing membranes to protons should disrupt oxidation and phosphoryltion (add detergent →holes in membrane→ no H+ gradient created = no pH change) 4. mito must contain a proton ddriven ATP synthase |
|
|
If add small amount of dilute HCl to a suspension of mito, which will occur? |
ATP will be synthesized (artifical H+ gradient created) |
|
|
P:O ratio |
4 H+ into ATP synthase required to make 1 high E bond (ATP) = 3 H+ into ATP synthase + 1 H+ to bring Pi NADH: 1 pair of e- = 10 H+ → 2.5 ATP (P:O 2.5) FADH2: 1 pair of e- = 6 H+ → 1.5 ATP (P:O 1.5) |
|
|
coupled nature of ATP synthesis and generation of proton gradient |
under normal conditions: disrupt one then disrupt the other |
|
|
Glycerol phosphate shuttle |
-gets cytosolic NADH e- into e- transport chain cytosolic glycerol-3-phosphate dehydrogenase: DHAP + NADH + H+ → NAD+ + Glycerol-3-phosphate mitochondrial glycerol-3-phosphate dehydrogenase: (embedded in membrane on cyoplasmic side) glycerol-3-phosphate + FAD → DHAP + FADH2 e- then transferred to CoQ → complex III -loss of potential: 1.5 ATP produced -skeletal mm. and brain mito use b/c faster -no cmpds transported into mito |
|
|
Malate Aspartate Shuttle |
overall equation: NADHout + NADH+in → NAD+out + NADHin -no E loss -liver -slow key enzymes: 1. malate DH (cyto and matrix isozymes) 2. glutamate aminotransferase (matrix) 3. aspartate aminotransferase (cyto) two transporters: 1. malate-α -ketoglutarate transporter 2. glutamate-aspartate transporter |
|
|
Malate Aspartate Shuttle: malate DHase |
(cytoplasm) OAA + NADH → NAD+ + malate → malate - α -ketoglutarate transporter → (matrix) malate + NAD+ → NADH + OAA |
|
|
Malate Aspartate Shuttle: glutamate aminotransferase |
OAA + glutamate → α - ketoglutarate + aspartate α - ketoglutarate → malate- α - ketoglutarate transporter → cytoplasm aspartate → glutamate-aspartate transporter → cytoplasm |
|
|
Malate Aspartate Shuttle: aspartate aminotransferase |
aspartate + α -ketoglutarate → glutamate + OAA glutamate → glutamate-aspartate transporter |
|
|
How to get cytoplasmic NADH into the mito for e- transport chain |
No NADH transporter in mito IM 1. malate aspartate shuttle (liver) 2. glycerol phosphate shuttle (mm., brain) |
|
|
uncouplers |
-cause membrane to become permeable to H+ → dissipate e- gradient → no ATP synthesis → respiration continues at increased rate in attempt to re-establish H+ gradient UCP1: thermogenin UCP2-5: unknown function (drug companies trying to create drugs to partially uncouple → weight loss) |
|
|
Thermogenin (UCP1) |
-natural uncoupler (babies, brown fat) -activated by Norepi -dissipation of proton gradient → heat production |
|
|
How oxidative phosphorylation can be inhibited |
1. block e- flow along the chain 2. block ATP synthase activity 3. allow free ion flow across inner mito membrane |
|
|
why does blocking ATP synthase block oxidation? |
enhanced H+ gradient |
|
|
Classes of ETC inhibitors |
1. Sites w/n ETC 2. ATP Synthase 3. Uncouplers 4. ATP/ADP exchange |
|
|
Rotenone |
Complex I blocked: specific inhibition of e- transfer from Fe-S to CoQ |
|
|
Antimycin A |
blocks complex III: CytC can't accept e- from it |
|
|
Cyanide |
inhibit complex IV: bind Fe in Cyts preventing e- transfer |
|
|
carbon monoxide |
inhibit complex IV: bind Fe in Cyts preventing e- transfer |
|
|
ETC inhibitors: sites w/n ETC |
a. Rotenone - I b. Amytal - I c. Antimycin A- III d CN, CO - IV |
|
|
ETC inhibitors: ATP synthase |
a. oligiomycin- block H+ entry b. DCCD- block H+ entry |
|
|
ETC inhibitors: Uncouplers |
a. CCCP - permeable to H+ b. DNP - permeable to H+ c. Valinomycin - permeable to K+ → destrs elec. grad d. Thermogenin - permeable to H+ |
|
|
ETC inhibitors: ATP/ADP exchange |
atractyloside- blocks ADP entry into mito → ATP synthase can't work |
|
|
Oxygen Consumption Graph and ETC inhibitors |
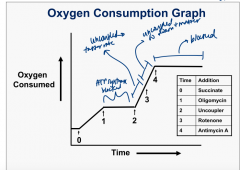
|
|
|
ADP entry into mito |
*ATP synthesis regulated by levels of available ADP, Pi Two transporters embedded in IM: *both driven by H+ gradient 1. Adenosine Nucleotide Translocase: antiporter ADP(3-) → matrix (→ ATP synthase) ATP (4-) → intermembrane space 2. Phosphate translocase: symporter H2PO4- and H+ → matrix (H2PO4- → ATP synthase) |
|
|
Oxidative Phosphorylation Regulation |
-by Energy Charge: (and O2) normal= 0.8 <0.8 → E production stimulated > 0.8 → ADP supplies in mito become rate limiting → ATP synthesis stops |
|
|
Energy Charge |

(represents # high E phosphate bonds) |
|
|
Energy yield from Glucose Oxidation: Glycolysis |

|
|
|
Energy yield from Glucose Oxidation: PDH and TCA |

|
|
|
Energy yield from Glucose Oxidation: Glucose → CO2 and H2O |
Glycerol-phosphate shuttle: 30 ATP malate-aspartate shuttle: 32 |
|
|
OXPHOS diseases arising form mtDNA mutations |
-affect organs w/ high E requirements = NS and mm. -partial activity loss in complexes I, III, IV → exercise intolerance -range of symptoms 1. deletions of mtDNA 2. Point mutations in tRNA or rRNA genes 3. missense mutations |
|
|
OXPHOS diseases arising form mtDNA mutations: deletions of mtDNA |
1. Kearns-Sayre syndrome (nerve and mm.) 2. Pearson Syndrome (bone marrow) |
|
|
OXPHOS diseases arising form mtDNA mutations: point mutations in tRNA or rRNA genes |
a. MERRF (myoclonic epilepsy and ragged red fiber disease): tRNA lys b. MELAS (mitco encephalopathy, lactic acidosis and strokelike episodes): 80% tRNA leu |
|
|
OXPHOS diseases arising form mtDNA mutations: missense mutations |
1. Lehigh disease (nerve and mm., 25% in ATP stnthase) 2. LHON (leber hereditary optic neuropathy): 90% complex I |
|
|
TCA cycle and cancer |
mutations in certain enzymes (cyto isoform) → tumer production: 1. succinate DH → succinate accummulation -familial paraganglioma, NS -loss of function 2. Fumarase → fumarate accumulation -autosomal dom.: --cutaneous and uterine leiomyomas --hereditary leiomylomatosis and renal cancer syndrome (age of onset 25-30) -autosomal recessive: --fumarate deficiency: progressive encephalopathy, cerebral strophy, seizures, hypotonia, renal developmental delay -loss of function -enzyme is homotetramer 3. isocitrate DH: gain of function mutation so can make 2-hydroxyglutarate instead of α -ketoglutarate -gliomas, acute myeloid leukemia **accumulation of key metabolites → altered gene expression and O2 sensing → tumor formation |
|
|
α -ketoglutarate and cancer |
-generated by IDH3 --also used for hydroxylation reactions: prolyl hydroxylase, collagen formation, N-methyl lysine hydrozylase = methylated histones, methy cytosine demethylase (genes w/ methyl C inactived) -O2 + α -KG → succinate + CO2 + hydroxylated product |
|
|
generation of α -KG, α -KG and cancer |
three isozymes: IDH1- cyto, NADP+ IDH2- mito, NADP+ IDH3- mito, TCA, linked to NAD+ mutation of of Arg at active site of IDH1/2: α -KG + NADPH → 2-hydroxyglutarate + NADP+ -mutation increases affinity for α -KG and decreases affinity for isocitrate at active site |
|
|
inhibition of N-methyl lysine hydroxylase (histone demethylases) |
-methylation of histone tail on lys → activation/inactivation of gene normally: α -KH binds hydroxylase → converts methyl to hydroxymethyl → lost as formaldehyde, N loses a methyl -2-hydroxyglutarate bins to active site of hydroxylase instead of α -KG → can't convert methyl to hydroxymethyl → alteration of gene expression |
|
|
what blocks histone demethylases |
IDH mutations, SDH muts, fumarase muts → buikd up of succinate (or fumarate→ succinate) → product inhibition → inability to demethylate histones → alteration of gene expression during differentiation |
|
|
inhibition of methylcytosine dioxygenase |
α -KG dependent enzyme hydroxylates methyl group on cyt in promoter regions → removal IDH, SH, FH muts: inhibition of hydroxylation → genome hypermethylation → altered gene expression and differentiation causes: acute myeloid leukemia (AML)- methylation pattern of RBC genome resembles stem cell |
|
|
hypoxia inducible factor (HIF) |
Normal cellular O2 levels: HIF proline hydroxylation by prolyl hydroxylase→ ubiquitin added → degraded by proteasome accummulation of 2-hydroxyglutarate, succinate, or fumarate → blocked hydroxylation by prolyl hydroxylase→ HIF active → increased glyvolysis and other events → cancer cell |
|
|
treating TCA cycle enzyme tumors |
block 2-hydroxyglutarate formation: 1. IDH-1 inhibitor: → reversed histone meth, slowed growth, promoted differentiation, didnt affect methylytosine demeth 2. IDH-2 inhibitor: →> reversed DNA and histone hypermeth amd induced differentiation |
|

|
MELAS (mutated tRNA leu) |