Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
129 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Describe DNA methylation.
What is its purpose |
Template strand cytosine and adenine are methylated in DNA replication, which allows MMR enzymes to distinguish btw old and new strands
"Methylation makes DNA Mute" |
|
|
|
Def: Hypermethylation
|
Inactivates transcription of DNA
|
|
|
|
Histone acetylation
|
Relaxes DNA coiling, allowing for transcription
"Acetylation makes DNA ACTIVE" |
|
|
|
Which bond btw DNA nucleotides is stronger?
What is the significance of this property? |
G-C bond has 3 H-bonds whereas A-T bond has 2 H-bonds.
Increased GC content = increased melting temp |
|
|
|
Amino acids necessary for purine synthesis
|
"GAG"
Glycine, Aspartate, Glutamine |
|
|
|
MOA: Hydroxyurea, 6-MP, 5-FU, MTX, TMP
|

HU: inhibits ribonucleotide reductase
6-MP: blocks de novo purine synthesis 5-FU: inhibits thymidylate synthase (decrease dTMP) MTX: inhibits DHFR (decrease dTMP) TMP: inhibits bacterial dihydrofolate reductase (decrease dTMP) |
|
|
|
Orotic aciduria MOA
Findings? Tx? |
Inability to convert orotic acid to UMP (de novo pyrimidine synthesis pathway) due to defect in either orotic acid phosphoribosyltransferase or orotidine 5'-phosphate decarboxylase. Autosomal recessive
Findings: increased orotic acid in urine, megaloblastic anemia (doe not improve with administration of Vit B12 or folate), failure to thrive. No hyperammonemia (vs OTC deficiency - increased orotic acid with hyperammonemia Tx: oral uridine administration |
|
|
|
How does Orotic aciduria differ from OTC deficiency
|
No hyperammonemia (vs OTC deficiency - increased orotic acid with hyperammonemia)
|
|
|
|
Adenosine deaminase deficiency MOA
|
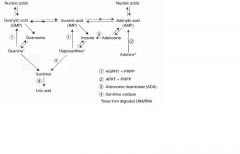
Excess ATP or dATP imbalances nucleotide pool via feedback inhibition of ribonucleotide reductase = prevents DNA synthesis and decreases lymphocyte count
One of the major causes of SCID |
|
|
|
Lesch-Nyhan syndrome MOA
Findings? |
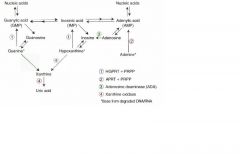
Defective purine salvage owing to absence of HGPRT which converts hypoxanthing to IMP and guanine to GMP. Results in excess uric acid production and de novo purine synthesis.
Findings: retardation, self-mutilation, aggression, hyperuricemia, gout, choreoathetosis X-linked recessive |
|
|
|
Discuss the four mutations in DNA: silent, missense, nonsense, and frame shift
|
Silent: same AA, often base change in 3rd position of codon (tRNA wobble)
Missense: Changed AA (conservative - new AA is similar in chemical structure) Nonsense: change resulting in early stop codon "stop the nonsense!" Frame shift: change resulting in misreading of all nucleotides downstream, usually resulting in a truncated, nonfunctional protein. |
|
|
|
DNA topoisomerase: MOA
Which medication inhibits this enzyme? |
Creates a nick in the helix to relieve supercoils created during replication.
Fluoroquinolones - inhibit DNA gyrase (prokaryotic topoisomerase II) |
|
|
|
Fxn of:
Primase DNA pol III DNA pol I |
Primase: makes an RNA primer on which DNA pol III can initiate replication
DNA pol III: Prokaryotic only. 5' --> 3' synthesis and proofreads with 3' --> 5' exonuclease DNA pol I: Prokaryotic only. Degrades RNA primer and fills in the gap with DNA. (DNA pol I excises RNA primer with 5' --> 3' exonuclease. |
|
|
|
DNA repair mechanisms: NER, BER, MMR, NHEJ
|
NER: specific endonucleases release the oligonucleotide-containing damaged bases; DNA pol and ligase fill and reseal the gap.
BER: specific glycosylases recognize and remove damaged bases, AP endonuclease cuts DNA at apyrimidinic site, empty sugar is removed, and the gap is filled and resealed. MMR: unmethylated, newly synthesized string is recognized, mismatched nucleotides are removed, and the gap is filled and resealed. NHEJ: Brings together two ends of DNA fragments. No requirement for homology. Q: Name a condition that occurs with defects in each of the repair pathways |
NER: Xeroderma pigmentosum, which prevents repair of thymidine dimers due to UV light exposure
BER: imp in repair of spontaneous/toxic deamination MMR: HNPCC NHEJ: Ataxia telangiectasia |
|
|
DNA/RNA/protein synthesis direction
|
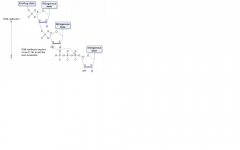
DNA and RNA are both synthesized in the 5' --> 3' direction.
5' of the incoming nucleotide bears the triphosphate (energy source for the bond). The triphosphate bond is the target of the 3' hydroxyl attack. Drugs blocking DNA replication often have modified 3' OH, preventing addition of the next nucleotide (aka "chain termination" mRNA is read 5' to 3' Protein synthesis is N to C |
|
|
|
Types of RNA
|
rRNA is the most abundant type
mRNA is the longest type tRNA is the smallest type "Rampant, Massive, Tiny" |
|
|
|
Start and stop codons
|
start: AUG (methionine in eukaryotes and forml-methionine (f-Met) in prokaryotes)
stop: UGA, UAA, UAG UGA = U Go Away UAA = U Are Away UAG = U Are Gonee |
|
|
|
Regulation of gene expression: Define Enhancer and Silencer
Where are each of these typically located? |
Enhancer: Stretch of DNA that alters gene expression by binding transcription factors
Silencer: Site where negative regulators (repressors) bind. Location: both may be located close to, far from, or even within (in an intron) the gene whose expression it regulates |
|
|
|
RNA polymerases in Eukaryotes and Prokaryotes - What do each of the RNA pol types do?
|
Eukaryotes- RNA pol I makes rRNA, RNA pol II makes mRNA, and RNA pol III makes tRNA.
No proofreading function, but can initiate chains. RNA pol II opens DNA at promoter site. Prokaryotes- one RNA pol (multisubunit complex) makes all 3 types of RNA Q: alpha-amantin --> what does it do, where is it found, and what does it cause? |
alpha amantin is found in death cap mushrooms. It inhibits RNA pol II, causing liver failure if ingested
|
|
|
lkjklj
|
ljklj
|
|
|
|
asf
|
sdf
|
|
|
|
RNA processing (eukaryotes)
|
Initial transcript is called heterogenous nuclear RNA (hnRNA) destined for translation is called pre-mRNA.
Processing occurs in nucleus. After transcription: 1) Capping on 5' end (addition of 7-methylguanosine occurs in cytosol) 2) polyadenylation on 3' end (=200 As) 3) splicing out of introns Capped, tailed, and spliced transcript is called mRNA Only processed RNA is transported out of the nucleus |
|
|
|
Discuss the regulation of the lac operon
|
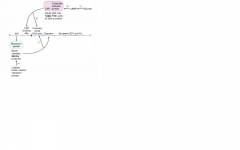
When active, E. coli can metabolize lactose. Lactose inhibits the repressor. Glucose inhibits the activator.
|
|
|
|
Discuss tRNA charging
|
Aminoacyl-tRNA synthetase (1 per aa, "matchmaker," uses ATP) scrutinizes aa before and after it binds to tRNA. If incorrect, bond is hydrolyzed.
The aa-tRNA bond has energy for formation of peptide bond. A mischarged tRNA reads usual codon but inserts wrong aa. Which medication prevents accurate tRNA charging? |
Tetracyclines bind 30S subunit, preventing attachment of aminoacyl-tRNA.
|
|
|
Discuss steps of protein synthesis (as it occurs in the cytoplasm from mRNA)
|

Initiation: activated by GTP hydrolysis, initiation factors help assemble the 40S ribosomal subunit with the initiator tRNA and are released when the mRNA and the ribosomal subunit assemble with the complex.
Elongation: 1) aminoacyl-tRNA binds to A site (except for initiator methionine) 2) Ribosomal rRNA (aka "ribozyme") catalyzes peptide bond formation, transfers growing polypeptide to amino acid in A site. 3) Ribosome advances 3 nucleotides toward 3' end of RNA, moving peptidyl RNA to P site (translocation) Q: Protein synthesis inhibitors |
Aminoglycosides: bind 30S and inhibit formation of the initiation complex and cause misreading of mRNA.
Chloramphenicol: inhibits 50S peptidyltransferase Macrolides: block translocation Clindamycin and Chloramphenicol: block peptide bond formation |
|
|
When is ATP and GTP used in the formation of protein?
|
ATP- tRNA Activation (charging)
GTP- tRNA Gripping and Going places (translocation) |
|
|
|
Regulation of cell cycle (4 proteins/complexes)
|
CDK: Cyclin-dependent kinases; constitutive and inactive
Cyclins: regulatory proteins that control cell cycle events; phase specific; activate CDKs Cyclin-CDK complexes: must be both activated and inactivated for cell cycle to progress Tumor suppressors: Rb and p53 normally inhibit G1-to-S progression; mutations in these genes result in unrestrained growth |
|
|
|
Cell types: Permanent, Stable (quiescent), Labile
|
Permanent: remain in G0, regnerate from stem cells
Stable: Enter G1 from G0 when stimulated Labile: Never go to G0, divide rapidly with a short G1 Which cell types are considered permanent, stable, or labile? |
Permanent: neurons, skeletal and cardiac muscle, RBCs
Stable: hepatocytes, lymphocytes Labile: Bone marrow, gut epithelium hair follicles |
|
|
RER: function, Nissl bodies, Free ribosomes
Which cells are rich in RER? |
Site of synthesis of secretory (exported) proteins and of N-linked oligosaccharide addition to many proteins
Nissle bodies (RER in neurons) synthesize enzymes and peptide neurotransmitters Free ribosomes - unattached to any membrane; site of synthesis of cytosolic and organellar proteins Cells rich in RER: mucus-secreting goblet cells of small intesting + antibody-secreting plasma cells |
|
|
|
SER: function
Which cells are rich in SER? |
Site of steroid synthesis and detox of drugs and poisons
Cells rich in SER: liver hepatocytes and steroid hormone-producing cells of the adrenal cortex |
|
|
|
Cell Trafficking: Golgi distribution center
|
Finish this??
|
|
|
|
Peroxisome definition
|
Membrane enclosed organelle involved in catabolism of very long fatty acids and amino acids
|
|
|
|
Drugs that act on microtubules:
|
1) Mebendazole/thiabendazole (antihelminthic)
2) Griseofulvin (antifungal) 3) Vincristine/vinblastine (anti-cancer) 4) Paclitaxel (anti-breast cancer) 5) Colchicine (anti-gout) |
|
|
|
Chediak-Higashi Syndrome: MOA and clinical findings
|
Microtubule polymerization defect resulting in decreased fusion of phagosomes and lysozomes.
Results in recurrent pyogenic infections, partial albinism, and peripheral neuropathy |
|
|
|
Molecular motor proteins: Kinesin and Dynein
Describe movement across microtubules. |
Kinesin moves anterograde from negative to positive end
Dynein moves retrograde from positive to negative end ("dying") |
|
|
|
Describe structure of microtubules
|
Cylindrical structure composed of a helical array of polymerized dimers of alpha and beta tubulin.
|
|
|
|
Kartagener's syndrome: MOA and clinical findings
|
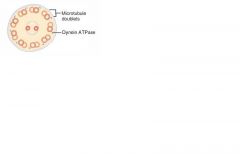
MOA: immotile cilia due to a dynein arm defect.
Clinical findings: male and female infertility (sperm immotile), bronchiectasis, and recurrent sinusitis (bacteria and particles not pushed out); associated with situs inversus (organs are mirrored) |
|
|
|
Cytoskeletal elements and their functions
|
Actin and myosin: microvilli, muscle contraction, cytokinesis, adherens junctions
Microtubule: Movement (cilia, flagella, mitotic spindle, axonal trafficking, centrioles) Intermediate filaments: Structure (vimentin, desmin, cytokeratin, lamins, GFAP, neurofilaments) |
|
|
|
Immunohistochemical stains for intermediate filaments
|
Vimentin: Connective tissue
Desmin: muscle tissue Cytokeratin: epithelial cells GFAP: NeuroGlia Neurofilaments: neurons |
|
|
|
Sodium pump: name some drugs that interfere with its functioning
|
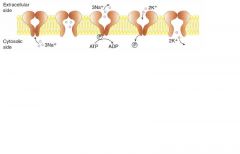
Ouabain: inhibits by binding to K+ site
Cardiac glycosides (digoxin and digitoxin): directly inhibit hte Na+/K+ ATPase, which leads to indirect inhibition of Na+/Ca2+ exchange. increased intracellular Ca2+ = increased cardiac contractility |
|
|
|
Collagen: Explain different types and what diseases may occur with each defective type.
|
Be (So Totally) Cool, Read Books
Type 1 (90%): Bone, Skin, Tendon; defective in osteogenesis imperfecta Type II: Cartilage (CarTWOlage) Type III: Reticulin - skin, blood vessels, uterus, fetal tissue, granulation tissue (defective in Ehlers-Danlos (ThreE D) Type IV: Basement membrane or basal lamina ("Type 4 under the floor," defective in Alport syndrome) |
|
|
|
Osteogenesis imperfecta
|
Genetic bone disorder (brittle bone dz) caused by a variety of gene defects
Most common form is autosomal dominant with abnormal type 1 collagen, causing 1) multiple fractures with minimal trauma (may occur during birth process), 2) blue sclerae due to the translucency of the connective tissue over the choroid, 3) hearing loss due to abnormal middle ear bones, 4) dental imperfections due to lack of dentin Other: may be confused with child abuse; type II is fatal in utero in the neonatal period |
|
|
|
Ehlers-Danlos syndrome
|
Faulty collagen synthesis causing: 1) hyperextensible skin 2) tendency to bleed (easy bruising) 3) hypermobile joints
May be associated with joint dislocation, berry aneurysms, organ rupture Type III collagen most frequently affected (Type ThreE D) |
|
|
|
Alport syndrome
|
Due to a variety of gene defects resulting in abnormal type IV collagen.
Most common form is X linked recessive. Characterized by progressive hereditary nephritis and deafness. May be associated with ocular disturbances. Type IV collage is an important structural component of the basement membrane of the kidney, ears, and eyes |
|
|
|
Elastin: describe this protein and defects that occur due to defects
|
Stretchy protein within lungs, large arteries, elastic ligaments, vocal cords, ligamenta flava (connect vertebrae --> relaxed and stretched conformations)
Broken down by elastase, which is normally inhibited by alpha 1 antitrypsin Marfan syndrome: caused by a defect in fibrillin Emphysema or cirrhosis: can be caused by alpha 1 antitrypsin deficiency, resulting in excess elastase activity. (side: wrinkles of aging are due to reduced collagen and elastin production) |
|
|
|
Describe how to clone a DNA molecule into bacteria
|
Cloning is the production of a recombinant DNA molecule that is self-perpetuating.
1) Isolate eukaryotic mRNA (post-RNA processing steps) of interest 2) Expose mRNA to RT to produce cDNA 3) Insert cDNA fragments into bacterial plasmids containing antibiotic resistance genes 4) Surviving bacteria on antibiotic medium produce cDNA library |
|
|
|
Loss of heterozygosity: definition + example
|
If a patient inherits or develops a mutation in a tumor suppressor gene, the complementary allele must be deleted/mutated before cancer develops. This is not true of oncogenes.
Ex: retinoblastoma |
|
|
|
Dominant negative mutation: definition + example
|
Exerts a dominant effect. A heterozygote produces a nonfunctional altered protein that also prevents the normal gene product from functioning.
Ex: mutation of Tx factor in its allosteric site. Nonfunctioning mutant can still bind DNA, preventing WT Tx factor from binding |
|
|
|
Linkage disequilibrium
|
Tendency for certain alleles at 2 linked loci to occur together more often than expected by chance. Measured in a population, not in a family, and often varies in different populations.
|
|
|
|
Mosaicism: definition + example
|
Occurs when cells in the body differ in genetic makeup due to postfertilization loss of genetic information during mitosis.
Can be germ-line mosaic (gonadal mosaicism), which may produce dz that is not carried by parent's somatic cells. Ex: Lyonization- random X inactivation of females; mutation in the embryonic precursor of the bone marrow stem cell --> a hematologic mosaic individual; a chimeric individual is derived from 2 zygotes that subsequently fuse. |
|
|
|
Locus heterogeneity: definition + example
|
Mutations at different loci can produce the same phenotype
Ex: Marfan's syndrome, MEN 2B, and homocystinuria; all cause marfanoid habitus; albinism |
|
|
|
Heteroplasmy: definition
|
Presence of both normal and mutated mtDNA, resulting in variable expression in mitochondrial inherited dz
|
|
|
|
Uniparental disomy: definition
|
Offspring receives 2 copies of a chromosome from 1 parent and no copies from the other parent
|
|
|
|
Hardy Weinberg population genetics: frequency of homozygosity, frequency of heterozygosity, frequency of X-linked recessive dz in males and in females.
|
p^2 + 2pq + q^2 = 1, and p + q =1
frequency of homozygosity for allele p = p squared 2pq = frequency of heterozygosity (carrier frequency) Frequency of X-linked recessive dz in males = q and in females = q squared Q: What does Hardy-Weinberg law assume? |
1) No mutation occurring at the locus
2) No selection for any of the genotypes at the locus (no selective advantage) 3) Completely random mating 4) No migration |
|
|
Imprinting: definition
|
At some loci, only 1 allele is active; the other is inactive (imprinted/inactivated by methylation). With 1 allele inactivated, deletion of the active allele --> disease
|
|
|
|
Prader-Willi syndrome: def and findings
|
Individual has normally inactivated maternal allele. PATERNAL allele should be active but is deleted.
Clinical findings: Mental retardation, hyperphagia, obesity, hypogonadism, hypotonia Both Prader-Walli and AngelMan's syndromes are due to inactivation or deletion of genes on chromosome 15. Can also occur as a result of uniparental disomy. |
|
|
|
AngelMan's syndrome: def and findings
|
Individual has normally inactivated paternal allele. MATERNAL allele should be active but is deleted.
Clinical findings: Mental retardation, seizures, ataxia, inappropriate laughter ("happy puppet") Both Prader-Walli and AngelMan's syndromes are due to inactivation or deletion of genes on chromosome 15. Can also occur as a result of uniparental disomy. |
|
|
|
Mitochondrial inheritance
|
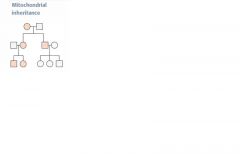
Transmitted only through mother. All offspring of affected females may show signs of dz. Often due to failures in oxidative phosphorylation.
Variable expression in population due to heteroplasmy. Mitochondrial myopathies- Leber's hereditary optic neuropathy (acute loss of central vision), myoclonic epilepsy, and mitochondrial encephalopathy. Characterized by "ragged red fibers" on microscopy. |
|
|
|
Achondroplasia
|
Autosomal Dominant
Cell signaling defect of fibrobblast growth factor (FGF) receptor 3. Sx: Dwarfism; short limbs, but head and trunk are normal size. Associated with advanced paternal age. |
|
|
|
Autosomal dominant polycystic kidney dz (ADPKD)
|
Autosomal Dominant
ALWAYS bilateral, massive enlargement of kidneys due to multiple large cysts. 90% of cases due to mutation in PKD1. Sx: flank pain, hematuria, HTN, progressive renal failure Associated with polycystic liver dz, berry aneurysms, mitral valve prolapse. (Infantile form is recessive) |
|
|
|
Familial adenomatous polyposis
|
Autosomal Dominant
Mutations on chromosome 5 (APC gene) --> 5 letters in polyp Colon becomes covered with adenomatous polyps after puberty. Progresses to colon cancer unless resected. |
|
|
|
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome)
|
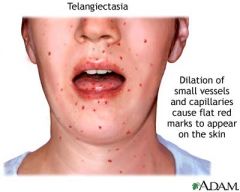
Autosomal Dominant
Inherited disorder of blood vessels. Sx: telangiectasia, recurrent epistaxis (nosebleed), skin discolorations, AVMs |
|
|
|
Hereditary spherocytosis
|
Autosomal Dominant
Spheroid erythrocytes due to spectrin or ankryin defect Sx: hemolytic anemia; increased MCHC Splenectomy is curative |
|
|
|
Huntington's dz
|
Autosomal Dominant
Gene located on chromosome 4 ("hunting 4 food"); trinucleotide repeat disorder (CAG)n Sx: depression, progressive dementia, choreiform movements, caudate atrophy, and decreased levels of GABA and ACh in the brain Sx manifest in affected individuals btw the ages of 20 and 50. |
|
|
|
Marfan's syndrome
|
Autosomal Dominant
Fibrillin gene mutation - connective tissue disorder affecting skeleton, heart, and eyes. Sx: tall with long extremities, pectus excavatum |
|
|
|
Multiple endocrine neoplasias (MEN)
|
Autosomal dominant
Several distinct syndromes (1, 2A, 2B) characterized by familial tumors of the endocrine glands, including those of the pancreas, parathyroid, pituitary, thyroid, and adrenal medulla. MEN2A and 2B are associated with ret gene |
|
|
|
Neurofibromatosis type 1
|
Autosomal dominant; On long arm of chromosome 17
Sx: cafe au lait spots, neural tumors, Lisch nodules (pigmented iris hamartomas). Also marked by skeletal disorders (eg scoliosis) and optic pathway gliomas. |
|
|
|
Neurofibromatosis type 2
|
Autosomal dominant - NF2 gene on chromosome 22; "type 2 = 22"
Bilateral acoustic schwannomas, juvenile cataracts |
|
|
|
Tuberous sclerosis
|
Autosomal dominant
Sx: facial lesions (adenoma sebaceum), hypopigmented "ash leaf spots" on skin, cortical retinal hamartomas, seizures, mental retardation, renal cysts and renal angiomyolipomas, cardiac rhabdomyomas, increased incidence of astrocytomas. Incomplete penetrance, variable penetration |
|
|
|
Von Hippel-Lindau dz
|
Autosomal dominant - deletion of VHL gene (tumor suppressor) on chromosome 3 (3p). Results in constitutive expression of HIF (transciption factor) and activation of angiogenic growth factors ("Von Hippel-Lindau = 3 words for chromosome 3")
Sx: hemangioblastomas of retina/cerebellum/medulla; about half o affected individual develop multiple bilateral renal cell carcinomas and other tumors. |
|
|
|
Cystic Fibrosis: Discuss pathogenesis, clinical presentations, diagnosis, and treatment
|
Autosomal recessive defect in CFTR gene on chromosome 7
CFTR channel actively secretes Cl- in lungs and GI tract and actively resorbs Cl- from sweat. Defective Cl- channel --> secretion of abnormally thick mucus that plugs lungs, pancreas, and liver --> recurrent pulm infections (Pseudomonas species and S. aureus), chronic bronchitis, bronchiectasis, pancreatic insufficiency (malabsorption and steatorrhea), nasal polyps, and meconium ileus in newborns. Mutation causes abnormal protein folding, resulting in degradation of channel before reaching cell surface. Infertility in males due to bilateral absence of vas deferens. Fat-soluble deficiencies (ADEK). Can present as failure to thrive in infancy. Most common lethal genetic dz in Caucasians Decreased concentration of Cl- ions in sweat test is diagnostic. Tx: N-acetylcysteine to loosen mucus plugs (cleaves disulfide bonds within mucus glycoproteins). |
|
|
|
Duchenne's muscular dystrophy
|
X-linked frame-shift mutation --> deletion of dystrophin gene --> accelerated muscle breakdown.
Dystrophin helps anchor muscle fibers, primarily in skeletal and cardiac muscle. Weakness begins in pelvic girdle muscles and progresses superiorly. Pseudohypertrophy of calf muscles due to fibrofatty replacement of muscle; cardiac myopathy. Use of Gowers' maneuver, requiring assistance of the upper extremities to stand up, is characteristic. Onset before 5 yrs of age. Diagnose muscular dystrophies by increasing CPK and muscle bx. |
|
|
|
Becker's muscular dystrophy
|
X-linked mutation dystrophin gene (no frameshift). Less severe than Duchenne's. Onset is adolescence or early adulthood.
Dystrophin helps anchor muscle fibers, primarily in skeletal and cardiac muscle. Diagnose muscular dystrophies by increasing CPK and muscle bx. |
|
|
|
Fragile X syndrome
|
X-linked defect affecting the methylation and expression of FMR1 gene.
2nd most common cause of genetic mental retardation (after Down syndrome) Sx: macroorchidism (enlarged testes), long face with a large jaw, large everted ears, autism, mitral valve prolapse. Trinucleotide repeat disorder (CGG)n Fragile X = eXtra large testes, jaw, ears |
|
|
|
Trinucleotide repeat expansion diseases
|
Huntington's disease, myotonic dystrophy, Friedreich's ataxia, fragile X syndrome.
"Try HUNTING for MY FRIED eggs (X)" Fragile X syndrome = (CGG)n Friedreich's ataxia = (GAA)n Huntington's dz = (CAG)n Myotonic dystrophy = (CTG)n "X-Girlfriend's First Aid Helped Ace My Test" May show genetic anticipation (dz severity increases and age of onset decreases in successive generations; germline expansion in females) |
|
|
|
Autosomal trisomies: Down syndrome (trisomy 21)
Sx, Pathogenesis/Epidemiology, results of Pregnancy Quad Screen + US |
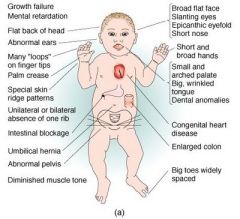
Sx: mental retardation, FLAT FACIES, prominent EPICANTHAL folds, SIMIAN CREASE, gap btw 1st 2 toes, duodenal atresia, congenital heart dz (most commonly septum primum-type ASD).
Associated with increased risk of ALL and Alzheimer's dz (>35yrs) 95% of cases due to meiotic nondisjunction of homologous chromosomes (associated with advanced maternal age; from 1:1500 in women less than 20 to 1:25 in women greater than 45) 4% cases due to robertsonian translocation. 1% of cases due to Down mosaicism (no maternal association) Pregnancy quad screen: decreased AFP, increased beta hCG, decreased estriol, increased inhibin A. US shows increased nuchal translucency. |
|
|
|
Edwards' syndrome
|
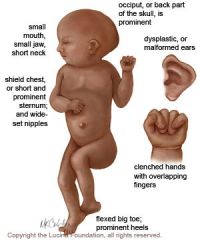
trisomy 18
Sx: severe mental retardation, rocker-bottom feet, micrognathia (small jaw), low set Ears, clenched hands, prominent occiput, congenital heart dz. Death usually occurs within 1 year of birth. Quad screen: decreased alpha fetoprotein, decreased Beta hCG, decreased estriol, normal inhibin A |
|
|
|
Patau's syndrome
|
trisomy 13
Sx: severe mental retardation, rocker-bottom feet, microphthalmia, microcephaly, CLEFT LIP/PALATE, HOLOPROSENCEPHALY, POLYDACTYLY, congenital heart disease. Dz usually occurs within one year of birth. Quad screen: normal alpha fetoprotein, normal beta hCG, normal estriol, normal inhibin A |
|
|
|
Robertsonian translocation
|
Nonreciprocal chromosomal translocation that commonly involves chromosome pairs 13, 14, 15, 21, and 22.
One of the most common types of translocations. Occurs when the long arms of 2 acrocentric chromsomes fuse at the centromere and the 2 short arms are lost. Balanced translocations normally do not cause any abnormal phenotype. Unbalanced translocations can result in miscarriage, stillbirth, and chromosomal imbalance (eg Down syndrome, Patau's syndrome) |
|
|
|
Cri-du-chat syndrome
|
Congenital microdeletion of short arm of chromosome 5 (46,XX or XY, 5p-)
Sx: microcephaly, moderate to severe mental retardation, high-pitched crying/mewing, epicanthal folds, cardiac abnormalities (VSD) |
|
|
|
Williams syndrome
|
Congenital microdeletion of long arm of chromosome 7 (deleted region includes the elastin gene)
Sx: distinctive "elfin" facies, mental retardation, hypercalcemia (increased sensitivity to Vit D), well-developed verbal skills, extreme friendliness with strangers, cardiovascular problems |
|
|
|
22q11 deletion syndromes
|
Variable presentation, including Cleft palate, Abnormal facies, Thymic aplasia --> T cell deficiency, Cardiac defects, Hypocalcemia secondary to parathyroid aplasia, due to microdeletion of chromosome 22q11
CATCH-22 Due to abberrant development of 3rd and 4th branchial pouches |
|
|
|
DiGeorge syndrome
|
22q11 deletion
thymic, parathyroid, and cardiac defects |
|
|
|
Velocardiofacial syndrome
|
22q11 deletion
Palate, facial, and cardiac defects |
|
|
|
Vitamins: fat soluble
Name and describe area of absorption |
ADEK.
Absorption dependent on gut (ileum) and pancreas. Toxicity more common than for water-soluble vitamins, because these accumulate in fat. |
|
|
|
Vitamins: water soluble
|
B1 (thiamin: TPP)
B2 (riboflavin: FAD, FMN) B3 (niacin: NAD+) B5 (pantothenic acid: CoA) B6 (pyridoxine: PLP) B12 (cobalamin) C (ascorbic acid) Biotin Folate All wash out easily from body except B12 and folate (stored in the liver). B-complex deficiencies often result in dermatitis, glossitis, and diarrhea. |
|
|
|
Vit A: deficiency and excess
|
Deficiency: night blindness, dry skin
Excess: arthralgias, fatigue, headaches, skin changes, sore throat, alopecia. Teratogenic (cleft palate, cardiac abnormalities), so a pregnancy test must be done before isotretinoin is prescribed for severe acne. |
|
|
|
Deficiency: night blindness, dry skin
Name vitamin |
Vitamin A
|
|
|
|
Excess: arthralgias, fatigue, headaches, skin changes, sore throat, alopecia. Teratogenic (cleft palate, cardiac abnormalities), so a pregnancy test must be done before isotretinoin is prescribed for severe acne.
Name vitamin |
Vitamin A
|
|
|
|
Vitamin B1 (thiamin):
Name function and deficiency |
In TPP, a cofactor for several enzymes in decarboxylation reactions:
1) pyruvate dehydrogenase (glycolysis) 2) alpha-ketoglutarate dehydrogenase (TCA cycle) 3) Transketolase (HMP shunt) 4) Branched-chain AA dehydrogenase Deficiency: Impaired glucose breakdown --> ATP depletion (glucose infusion can worsen); highly aerobic tissues (brain and heart) are affected first. Wernicke-Korsakoff syndrome and beriberi. Seen in malnutrition as well as alcoholism (secondary to malnutrition and malabsorption) |
|
|
|
Wernicke-Korsakoff
|
triad: confusion, ophthalmoplegia, ataxia
+ confabulation, personality change, permanent memory loss. Damage to medial dorsal nucleus of thalamus, mammillary bodies |
|
|
|
Beriberi
|
Vit B1 deficiency (Ber1Ber1)
Dry beriberi - polyneuritis, symmetrical muscle wasting Wet beriberi - high output cardiac failure (dilated cardiomyopathy), edema |
|
|
|
Vitamin B2: function and deficiency
|
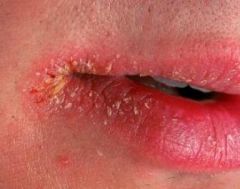
(riboflavin)
Function: cofactor in oxidation and reduction (eg FADH2) "FAD and FMN are derived from riboFlavin" (B2 = 2ATP) Deficiency: Cheilosis (inflammation of lips, scaling and fissures at corners of the mouth, corneal vascularization |
|
|
|
Deficiency of this vitamin leads to: Cheilosis (inflammation of lips, scaling and fissures at corners of the mouth, corneal vascularization
|
Vit B2 (riboflavin)
|
|
|
|
Vitamin B3 (niacin): function, deficiency, excess
|
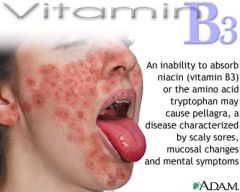
Function: Constituent of NAD+, NADP+ (used in redox reactions). Derived from tryptophan. Synthesis requires vitamin B6.
"NAD derived from Niacin" (B3 = 3 ATP) Deficiency: Glossitis. Severe deficiency leads to pellagra, which can be caused by Hartnup dz (decreased tryptophan metabolism), and INH (decreased vitamin B6). "The 3D's of B3: Diarrhea, Dermatitis, Dementia" Excess: Facial flushing (due to pharmacologic doses for treatment of hyperlipidemia). |
|
|
|
Vitamin B5 (pantothenate): function and deficiency
|
Function: essential component of CoA (a cofactor for acyl transfers) and fatty acid synthase
Deficiency: Dermatitis, enteritis, alopecia, adrenal insufficiency |
|
|
|
Deficiency: Dermatitis, enteritis, alopecia, adrenal insufficiency
|
Vit B5 deficiency (pantothenate)
|
|
|
|
Deficiency: Glossitis. Severe deficiency leads to pellagra, which can be caused by Hartnup dz (decreased tryptophan metabolism), and INH (decreased vitamin B6).
|
Vit B3 deficiency
"The 3D's of B3: Diarrhea, Dementia, Dermatitis" |
|
|
|
Vit B6: function and deficiency
|
pyridoxine
Function: Converted to pyridoxal phosphate, a cofactor used in transamination (eg ALT and AST), decarboxylation reactions, glycogen phosphorylase. Synthesis of cystathionine, heme, niacin, and GABA. Deficiency: Convulsions, hyperirritability, peripheral neuropathy (deficiency inducible by INH nad oral contraceptives), sideroblastic anemias due to impaired Hb synthesis an Fe excess |
|
|
|
Deficiency: Convulsions, hyperirritability, peripheral neuropathy (deficiency inducible by INH nad oral contraceptives), sideroblastic anemias due to impaired Hb synthesis an Fe excess
|
Vit B6 (pyridoxine) deficiency
|
|
|
|
Vitamin B12: function and deficiency (+ causes of deficiency)
|
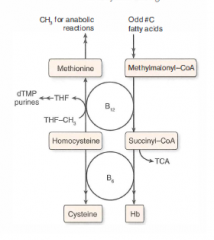
Function: cofactor for homocysteine methyltransferase (transfers CH3 groups as methylcobalamin) and methylmalonyl-CoA mutase
Deficiency: macrocytic, megaloblastic anemia, hypersegmented PMNs, neurologic sx (paresthesias, subacute combined degeneration) due to abnormal myelin. Prolonged deficiency leads to irreversible nervous system damage. Causes of deficiency: malabsorption (sprue, enteritis, Diphyllobothrium latum), lack of intrinsic factor (pernicious anemia, gastric bypass surgery), or absence of terminal ileum (Crohn's dz) Use Schilling test to detect etiology of the deficiency. |
|
|
|
Deficiency: macrocytic, megaloblastic anemia, hypersegmented PMNs, neurologic sx (paresthesias, subacute combined degeneration) due to abnormal myelin. Prolonged deficiency leads to irreversible nervous system damage.
|
Vit B12 (cobalamin) deficiency
Use Schilling test to detect etiology of the deficiency |
|
|
|
Vit C (ascorbic acid): function, deficiency, excess
|
Function: Antioxidant.
1) facilitates iron absorption by keeping iron in Fe2+ reduced state. 2) necessary for hydroxylation of proline and lysine in collagen synthesis 3) necessary for dopamine beta-hydroxylase, which converts DA to NE Deficiency: scurvy: swollen gums, bruising, hemarthrosis (bleeding into joints), anemia, poor wound healing. Weakened immune response. "Vitamin C deficiency causes sCurvy due to a Collagen synthesis defect" Excess: N/V, diarrhea, fatigue, sleep problems. Can increase risk of iron toxicity in predisposed individuals (transfusion, hereditary hemochromatosis) |
|
|
|
Deficiency: scurvy: swollen gums, bruising, hemarthrosis (bleeding into joints), anemia, poor wound healing. Weakened immune response.
|
Vit C deficiency
"Vitamin C deficiency causes sCurvy due to a Collagen synthesis defect" |
|
|
|
Vit D: function, deficiency, and excess
|
Function: increase intestinal absorption of calcium and phosphate, increase bone resorption.
25-OH D3=storage form 1,25-(OH)2 D3 (calcitriol) = active form Deficiency: rickets in children (bending bondes), osteomalacia in adults (soft bones), hypocalcemic tetany. Breast milk has decreased Vit D (supplement in dark-skinned patients) Excess: hypercalcemia, hypercalciuria, loss of appetite, stupor. Seen in sarcoidosis (increased activation of vit D by epithelioid macs) |
|
|
|
Vit E: function and deficiency
|
Function: Antioxidant (protects erythrocytes and membranes from free-radical damage).
"E is for erythrocyes" Deficiency: increased fragility of erythrocytes (hemolytic anemia), muscle weakness, posterior column and spinocerebellar tract demyelination |
|
|
|
Deficiency: increased fragility of erythrocytes (hemolytic anemia), muscle weakness, posterior column and spinocerebellar tract demyelination
|
Vit E deficiency
|
|
|
|
Vit K: function and deficiency
|
Function: catalyzes gamma carboxylation of glutamic acid residues on various proteins concerned with blood clotting. Synthesized by intestinal flora.
"K is for Koagulation" necessary for the synthesis of clotting factors 1972 and protein C and S Warfarin = vitamin K antagonist Deficiency: Neonatal hemorrhage with increased PT and increased aPTT but normal bleeding time (neonates have sterile intestines and are unable to synthesize vit K). Can also occur after prolonged use of broad-spectrum antibiotics. |
|
|
|
Zinc: function and deficiency
|
Function: essential for the activity of 100+ enzymes. Important in the formation of zinc fingers (transcription factor motif).
Deficiency: delayed wound healing, hypogonadism, decreased adult hair (axillary, facial, pubic), dysgeusia, anosmia. May predispose to alcoholic cirrhosis. |
|
|
|
Ethanol metabolism: limiting reagent, kinetics of alcohol dehydrogenase
2 drugs implicated in ethanol metabolism |

|
|
|
|
Ethanol hypoglycemia: Mechanism
|

|
|
|
|
Malnutrition: Kwashiorkor vs Marasus
|
Kwashiorkor: protein malnutrition resulting in skin lesions, edema, liver malfunction (fatty change due to decreased apoliprotein synthesis). Clinical picture is small child with swollen belly
"protein deficient MEAL: Malnutrition, Edema, Anemia, Liver (fatty) Marasmus: energy malnutrition resulting in tissue and muscle wasting, loss of subcutaneous fat, and variable edema. "Marasmus results in Muscle wasting" |
|
|
|
Metabolism sites: Mitochondria, Cytoplasm, Both
|
Mitochondria: fatty acid oxidation (beta oxidation), acetyl-CoA production, TCA cycle, oxidative phosphorylation
Cytoplasm: Glycolysis, FA synthesis, HMP shunt, protein synthesis (RER), steroid synthesis (SER) Both: Heme Synthesis, Urea cycle, Gluconeogenesis ("HUGs take Two) |
|
|
|
Rate determining enzymes of metabolic processes
|
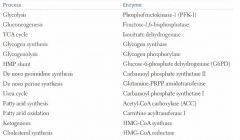
|
|
|
|
Glycolysis/ATP production: how many ATP produced by anaerobic glycolysis and aerobic metabolism in the heart, liver, and muscle
|
Aerobic metabolism of glucose produces 32 ATP via malate-aspartate shuttle (heart and liver), 30 ATP via glycerol-3-phosphate shuttle (muscle).
Anaerobic glycolysis produces only 2 net ATP per glucose molecule |
|
|
|
Universal electron acceptors
|
Nicotinamides (NAD+ from vitamin B3, NADP+) and flavin nucleotides (FAD+ from vitamin B2).
NAD+ is generally used in catabolic processes to carry reducing equivalents away as NADH. NADPH is used in anabolic processes (steroid and fatty acid synthesis) as a supply of reducing equivalents. NADPH is a product of the HMP shunt NADPH is used in: 1) anabolic processes 2) respiratory burst 3) P-450 4) Glutathione reductase |
|
|
|
Hexokinase vs. glucokinase
|
Phosphorylation of glucose to yield glucose-6-phosphate serves as hte first step of glycolysis (also serves as the first step of glycogen synthesis in the liver). Rxn is catalyzed by either hexokinase or glucokinase, depending on the location.
Hexokinase: ubiquitous. High affinity, (low Km), low capacity (low Vmax), uninduced by insulin; feedback inhibited by glucose-6-phosphate Glucokinase: Liver and beta cells of pancreas. Low affinity (high Km), high capacity (Vmax), induced by insulin (GLUcokinase is a GLUtton. It has high Vmax bc it cannot be satisfied. No direct feedback inhibition. Phosphorylates excess glucose (eg after a meal) to sequester it in the liver. Allows liver to serve as a blood glucose buffer. |
|
|
|
Glycolysis regulation, key enzymes
|

|
|
|
|
Regulation by F2,6 BP
|

|
|
|
|
Pyruvate dehydrogenase complex
|

|
|
|
|
Pyruvate dehydrogenase deficiency: Results and Sx, Tx
|
Causes backup of substrate (pyruvate and alanine), resulting in lactic acidosis. Can be congenital or acquired (as in alcoholics due to B1 deficiency)
Findings: neuological deficits Tx: increase intake of ketogenic nutrients (eg high fat content and increase lysine/leucine) Lysine and Leucine - the only purely ketogenic amino acids |
|
|
|
Functions of different pyruvate metabolic pathways
|
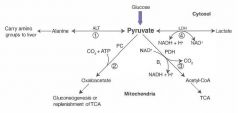
1) alanine carries AA groups to the liver from muscle
2) OAA can replenish TCA cycle or be used in gluconeogenesis 3) Transition from glycolysis to the TCA cycle 4) End of anaerobic glycolysis (major pathway in RBCs, leukocytes, kidney medulla, lens, testes, and cornea) |
|
|
|
TCA cycle (Krebs cycle)
|
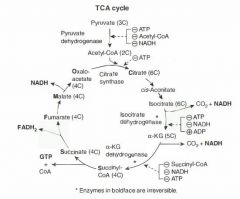
Pyruvate --> acetyl CoA produces 1 NADH, 1 CO2
TCA cycle produces 3 NADH, 1 FADH2, 2 CO2, 1 GTP per acetyl CoA = 12 ATP/acetyl-CoA (2x everything per glucose). TCA cycle reactions occur in the mitochondria. alpha-KG dehydrogenase complex requires the same cofactors as the pyruvate dehydrogenase complex (B1, B2, B3, B5, lipoid acid) "Citrate Is Krebs' Starting Substrate For Making Oxaloacetate" |
|
|
|
Discuss the ETC and oxidative phosphorylation.
|
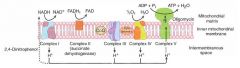
1) NADH electrons from glycolysis enter mitochondria via the malate-aspartate shuttle or glycerol-phosphate shuttle.
2) FADH2 electrons are transferred to complex II (at a lower energy level than NADH). 3) Passage of electrons results in the formation of a proton gradient that, coupled to oxidative phosphorylation, drives the production of ATP. |
|
|
|
Ox Phos poisons
|
1) Electron transport inhibitors: directly inhibit electron transport, causing a decrease of the protein gradient and block of ATP synthesis - rotenone, CN-, antimycin A, CO
2) ATP synthase inhibitors: Directly inhibit mitochondrial ATP synthase, causing an increased proton gradient. No ATP is produced because electron transport stops - oligomycin 3) Uncoupling agents: increased permeability of membrane, causing a decreased proton gradient and increased O2 consumption. ATP synthesis stops, but electron transport continues. Produces heat - 2,4-DNP, aspirin (fever often occur after aspirin overdose), thermogenin in brown fat. |
|
|
|
Gluconeogenesis, irreversible enzymes
|
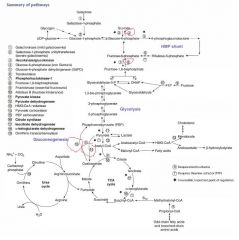
1) Pyruvate carboxylase
2) PEP carboxykinase 3) Fructose-1,6-bisphosphatase 4) Glucose-6-phosphatase |

|
|
|
HMP shunt (pentose phosphate pathway)
|

Provides a source of NADPH from an abundantly available glucose 6 phosphate (NADPH is required for reductive reactions, eg, glutathione reduction inside RBCs).
Additionally, this pathway yields ribose for nucleotide synthesis and glycolytic intermediates. 2 distinct phases (oxidative and nonoxidative), both of which occur in the cytoplasm. No ATP is used or produced Sites: lactating mammary glands, liver, adrenal cortex (sites of fatty acids or steroid synthesis), RBCs |
|
|
|
Respiratory burst (oxidative burst): discuss the role of NADPH oxidase and the condition that results from its deficiency
|
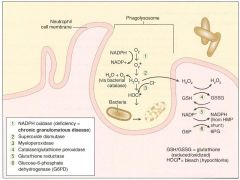
Involves activation of membrane-bound NADPH oxidase (eg in neutrophils, monocytes).
Plays an important role in the immune response --> rapid release of reactive oxygen intermediates (ROIs). Note that NADPH plays a role in the creation of ROIs and in their neutralization. WBCs of patients with CGD can utilize H2O2 generated by invading organisms and convert it to ROIs. Patients are at increased risk for infection by catalase-positive species (eg S. aureus, Aspergillus) bc they neutralize their own H2O2, leaving WBCs without ROIs for fighting infections. |
|
|
|
Glucose 6 phosphate dehydrogenase deficiency
|
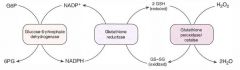
NADPH is necessary to keep glutathione reduced, which in turn detoxifies free radicals and peroxides.
Decreased NADPH in RBCs leads to hemolytic anemia due to poor RBC defense against oxidating agents (eg fava beans, sulfonamides, primaquine, antiTB drugs). Infection can also precipitate hemolysis (free radicals generated via inflammatory response can diffuse into RBCs and cause oxidative damage). Q: Discuss the disorder in terms of histopathology and pattern of inheritance. |
X-linked recessive disorder; most common human enzyme deficiency; more prevalent among blacks. Increases malarial resistance
Heinz bodies - oxidized Hemoglobin precipitated within RBCs Bite cells - result from the phagocytic removal of Heinz bodies by splenic macrophages. "Think Bite into some Heinz ketchup" |