Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
84 Cards in this Set
- Front
- Back
|
Define Hemoglobinopathy
|
STRUCTURAL defects in Hemoglobin resulting in an abnormal Hb molecule that can precipitate out/polymerize and lead to hemolysis
|
|
|
What is HbA?
|
Normal Hemoglobin with 2 Alpha & 2 Beta chains
|
|
|
What is HbA2?
|
Hb with 2 Alpha and 2 Delta chains
|
|
|
What is HbF?
|
Hb with 2 Alpha and 2 Gamma chains
HbF-AG (FAG) |
|
|
Hemoglobin whose gene is on Chromosome 16
|
Alpha
**you have 4 copies of Alpha genes |
|
|
Hemoglobin whose gene is on Chromosome 11
|
Beta, Gamma, Delta
**have 2 copies of each |
|
|
What is the abnormality in Sickle Cell Disease?
|
Point mutation in which Valine is substituted for Glutamate at position 6 of the BETA chain of Hemoglobin
|
|
|
Describe the Pathogenesis of Sickle Cell disease
|
1. Deoxygenation
2. Aggregation and polymerizatoin of HbS 3. Reversible sickling 4. Irreversible sickling 5. Hemolysis |
|
|
What type of hereditary disease is Sickle Cell Disease?
|
Autosomal Recessive = must have both recessive alleles to be diseased = HbS/HbS
|
|
|
What group of people have 8% of their population with the Sickle Cell trait (heterozygous = carry trait but are asymptomatic)?
|
African Americans
|
|
|
Describe the polymerization of Hb in Sickle Cell Disease
|
HbS polymerize only with other HbS and do so only in the Deoxy state
|
|
|
What conditions make Sickle Cell Disease worse?
|
1. Dehydration = increased concentration of RBC's with HbS
2. Low pH (acidic) = decreases Oxygen affinity for RBC --> causes Deoxy |
|
|
Describe the Sickle Cell Trait
|
Patients are Heterozygotes and only a portion of the hemoglobin is HbS and the remainder is normal HbA. RBC sickling and possibly hemolysis occur in hypoxia
|
|
|
List the pathologic findings in Sickle Cell Disease
|
1. Sickle Cells
2. Microvascular occlusion -> thrombosis and infarction due to Sickle cells stuck in small vessels 3. Autosplenectomy = due to repeated bouts of infarction 4. Bone Marrow Hyperplasia = due to hemolytic anemia 5. Extramedullary hematopoiesis = when BM cannot keep up with need 6. Gallstones = due to increased Bilirubin from breakdown of Heme from Hb |
|
|
Sickle Cell Disease
|
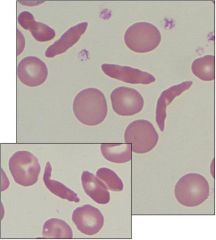
What disease is this?
|
|
|
Why is there pain in Sickle Cell Anemia?
|
Vaso-occlusive crises in the back or limbs due to microvasculature blockage by sickled cells
|
|
|
What would cause an Aplastic Crisis in Sickle Cell Anemia?
|
Parvovirus B19
|
|
|
What is the most common cause of death in Sickle Cell Anemia? Propose a possible mechanism
|
Infections
Mechanism: Autosplenectomy results in increased incidence of encapsulated organism infections -Strep pneumo & Hib -Salmonella -> Osteomyelitis |
|
|
What is the treatment for Sickle Cell Anemia? Explain the reasoning
|
Hydroxyurea
Increases the levels of HbF (gamma2 + delta2) while decreasing the levels of HbS |
|
|
What does Sickle Cell Trait protect against?
|
Falciparum malaria
|
|
|
Describe the distribution of normal adult hemoglobin
|
1. HbA = 96%
2. HbA2 = 3% (alpha2/delta2) 3. HbF = 1% (alpha2/gamma2) |
|
|
Define Thalassemias
|
Lack of or decreased synthesis of structurally normal hemoblogin chains
|
|
|
Describe the pathogenesis of Thalassemias
|
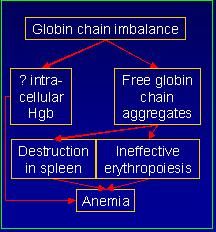
-
|
|
|
What ethnicities is Alpha-thalassemia most prevalent in?
|
Africa & SE ASIA
|
|
|
What ethnicities is Beta-thalassemia most prevalent in?
|
Africa
Asia MEDITERRANEAN |
|
|
In Thalassemias, What 2 things does Free globin chain aggregates result in?
|
1. Destruction in the spleen
2. Ineffective erythropoiesis = premature destruction of maturing erythroblasts within the BM |
|
|
What is Beta-Thalassemia and what is the cause?
|
1. Decreased synthesis of Beta chains
2. Point mutations causing: -Splicing errors (most common) -Promoter region (β+) -Chain termination (β0) **unlike Alpha-thalassemia, gene deletions are uncommon |
|
|
Resulting anemia in Beta-Thalassemia
|
Hypochormic, microcytic anemia
**b/c there is less Hb = less heme = less iron in RBC |
|
|
What Hb's are elevated in Beta-Thalassemia?
|
HbF (a2g2)
HbA2 (a2d2) |
|
|
Pathological findings of Beta-thalassemia
|
1. Bone Marrow Hyperplasia
-cortical thinning = "crewcut" skull x-ray =increased size of maxilla 2. Hepatosplenomegaly 3. Hemosiderosis - hemolysis is taking place in BM and Spleen where Fe+ is recoverable -Severe Thalassemias require blood transfusions |
|
|
Beta-Thalassemia
Target cells |
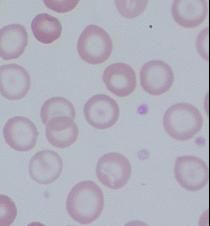
What disease?
What are the cells called? |
|
|
Beta-thalassemia
|
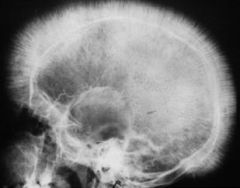
What disease causes "crewcut" skull on x-ray?
|
|
|
List the clinical features of Beta-Thalassemia Major
|
1. β0/β0 or β+/β+ = no functional Beta-globin
2. Severe anemia, ↑↑↑ HbF, ↑ HbA2 3. Transfusion dependent 4. Hemosiderosis 5. Treatment: BM transplant 6. Develops at 6 months of age when HbF levels decline β0/β0 = do not produce β-chains at all β+/β+ = reduced β-chain synthesis |
|
|
What is the genotype for β-Thalassemia intermedia
|
β0/β, β+/β+
**severe anemia, but not enough to require regular blood transfusions |
|
|
List the clinical features of β-Thalassemia minor
|
1. β0/β or β+/β = there is one normal Beta-globin gene
2. Asymptomatic 3. Mild anemia, ↑ HbF, ↑ HbA2 4. Protects against Falciparum malaria |
|
|
What is the Etiology of Alpha-Thalassemias?
|
GENE DELETIONS of alpha chains
**there are 2 alpha genes on each of our Chr. 16 = 4 total **can be various combos of deletions |
|
|
What is the silent carrier state of Alpha-Thalassemia?
|
1 out of 4 alpha genes is deleted (-a/aa)
- completely asymptomatic |
|
|
Describe the Alpha-Thalassemia Trait
|
2 deletions of the Alpha gene:
- (--/aa) or (-a/-a) |
|
|
What is the concern with individuals with the Alpha-Thalassemia Trait?
|
If 2 individuals with the genotype (--/aa) mate, they can produce a child with Hydrops Fetalis = Genetic Counseling
*25% chance of offspring with Hydrops fetalis |
|
|
Describe the HbH disease in Alpha-Thalassemia
|
3 deletions of Alpha chain (--/-a) causes increased HbH = tetramer of Beta chains (β4)
|
|
|
What is the result when there are 4 deletions of the Alpha gene in an individual?
|
Hydrops fetalis
|
|
|
Type of anemia in Alpha-Thalassemias
|
Hypochromic, microcytic anemia
|
|
|
What are the pathologic findings associated with Alpha-Thalassemia?
|
1. Hypochromic, microcytic anemia of variable severity
2. HbH disease 3. Bone Marrow Hyperplasia 4. Hepatosplenomegaly |
|
|
What is Barts Hemoglobin (Gamma4) associated with?
|
Hydrops fetalis Alpha-Thalassemia (lacks all 4 alpha-globin chains)
|
|
|
Alpha-Thalassemia (HbH disease = 3 deletions)
Target cells in upper Heinz bodies = HbH (β4) |
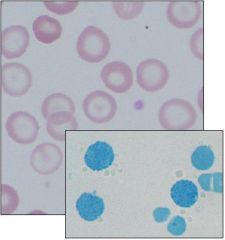
What disease is this?
How do you know? |
|
|
What ethnicity typically carries the cis genotype (--/αα)of the Alpha-Thalassemia trait?
|
Asians = if both partners have it there is a 25% chance of having child with intrauterine death
|
|
|
What ethnicity typically carries the trans genotype (a-/a-) of the Alpha Thalassemia trait?
|
African-Americans
|
|
|
Disease caused by an acquired deficiency of Glycosyl Phosphatidyl Inositol (GPI)-linked proteins due to mutations in Phosphatidylinositol Glycan A (PIGA)
|
Paroxysmal Nocturnal Hemoglobinuria
|
|
|
Describe the pathogenesis of Paroxysmal Nocturnal Hemoglobinuria
|
1. ↓ GPI anchored proteins (CD55, CD59, C8-binding protein, Decay Accelerating Factor)
2. ↑ complement sensitivity 3. Intravascular Hemolysis |
|
|
How do you diagnose Paroxysmal Nocturnal Hemoglobinuria?
|
Look for the presence of cell surface proteins, if missing it means PNH
|
|
|
What are the clinical findings of Paroxysmal Nocturnal Hemoglobinuria?
|
1. Anemia
2. Hemosiderinuria --> leads to iron deficiency 3. Venous thrombi which may be fatal |
|
|
What complications does Paroxysmal Nocturnal Hemoglobinuria cause an increased risk of? (3)
|
1. Aplastic anemia
2. Acute Leukemia 3. Venous thrombosis |
|
|
What cell type has the acquired defect in Paroxysmal Nocturnal Hemoglobinuria?
|
Multipotent Myeloid Stem Cell
|
|
|
Why would Paroxysmal Nocturnal Hemoglobinuria occur at night?
|
Respiratory acidosis, which occurs during slow breathing that causes retention of CO2, causes activation of Complement
|
|
|
What Screening test and what Confirmatory tests are used in Paroxysmal Noctural Hemoglobinuria?
|
Screening = Sucrose Hemolysis
- sucrose enhances complement destruction of RBC's Confirmatory = Acidified Serum Test (Ham test) - acidified serum activates the Alternative complement pathway |
|
|
What type of Anemias are Immunohemolytic anemias?
|
Extrinsic = environment is causing RBC destruction
|
|
|
Type of Antibody that causes Warm Autoimmune Hemolytic Anemia
|
IgG, active at 37' C
|
|
|
Describe the pathogenesis of Warm AIHA
|
1. IgG coated RBC
2. Fc receptor binds to Splenic Macrophages 3. Spherocytes 4. Destruction in the spleen = Extravascular |
|
|
What are the Secondary causes of Warm AIMA?
|
1. Lymphoproliferative disorders = Lymphoma = B cell neoplasm making Ab's against RBCs
2. Autoimmune diseases = SLE 3. Drugs -Haptens -Autoantibodies |
|
|
What drugs can act as Haptens and cause Warm AIHA?
|
1. Quinidine
2. Penicillin 3. Cephalosporin |
|
|
What drug can cause the production of an Autoantibody to RBC's causing Warm AIHA?
|
Methyldopa
**causes production of an Ab that cross-reacts with RBC |
|
|
What 2 disorders are characterized by Spherocytes and how do you differentiate?
|
1. Warm AIHA
2. Hereditary Spherocytosis **Warm AIHA will be Direct Coomb's test + = tests for Ab's coated on RBC's |
|
|
Type of Antibody that causes Cold Agglutinin Disease
|
IgM
|
|
|
Describe the Pathogenesis of Cold Agglutinin Disease
|
1. IgM coated RBC
2. RBC agglutination 3. Complement fixation 4. Intravascular and Extravascular Hemolysis |
|
|
Why does Cold Agglutinin Disease cause agglutination and complement fixation?
|
IgM is a pentamer so it can latch onto more than one RBC = agglutination
Agglutination causes Complement activation |
|
|
What are 2 acute causes of Cold Agglutinin Disease?
|
recovery from Mycoplasma pneumoniae
recovery from Infectious Mononucleosis |
|
|
What are teh 2 chronic causes of Cold Agglutinin Disease?
|
Idiopathic
Lymphoma |
|
|
Describe the Pathogenesis of Paroxysmal Cold Hemoglobinuria
|
IgG antibodies (cold hemolysins) bind to RBC at low temperature, fix complement, and cause hemolysis at temps above 30 C
|
|
|
What is the most important marker of Immune Hemolytic Anemias?
|
Direct Coombs test = direct antiglobulin test (DAT)
|
|
|
Warm AIHA
-Spherocytes **Spherocytes are also caused by Hereditary Spherocytosis |
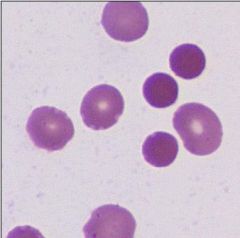
Autoimmune hemolytic anemia that would cause this
|
|
|
Cold Agglutinin Disease
-IgM causes agglutination of RBC's at temperatures lower than body temp |
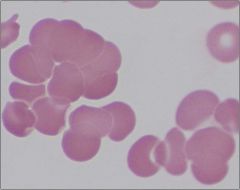
What is this disease?
|
|
|
What are the clinical features of Warm AIHA?
|
1. Variably severe anemia -> Splenomegaly
2. Treatment is directed to the underlying cause |
|
|
What are the clinical features of Cold Agglutinin Disease?
|
1. Variably severe anemia
2. Self limited 3. RAYNAUD PHENOMENON |
|
|
What is the clinical feature of Paroxysmal Cold Hemoglobinuria?
|
Intermittent massive hemolysis AFTER exposure to cold
|
|
|
Describe Macroangiopathic Hemolytic Anemia
|
RBC is hit against something which causes it to lyse
|
|
|
What are 2 possible causes of Macroangiopathic Hemolytic Anemia?
|
1. Aortic Stenosis
2. Prosthetic Heart Valves |
|
|
Describe Microangiopathic Hemolytic Anemia
|
partial occlusion of small vessels is the cause of mechanical disruption of the RBC's
|
|
|
Give 6 examples of causes of Microangiopathic Hemolytic Anemia
|
1. DIC
2. Thrombotic Thrombocytopenic Purpura 3. Hemolytic Uremic Syndrome 4. Malignant Hypertension 5. SLE 6. Disseminated Cancer |
|
|
What are the pathologic findings of Traumatic Hemolytic Anemia (Micro- or Macroantiopathic)
|
Schistocytes
|
|
|
Schistocytes
Micro- or Macroangiopathic Hemolytic Anemia = Trauma |
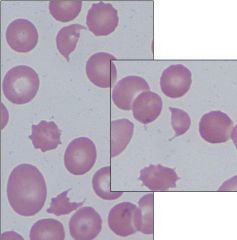
What are these cells called?
What is the cause? |
|
|
An 8-year-old AFRICAN AMERICAN boy presents complaining of SEVERE PAIN IN BOTH LEGS. The pain began after the boy attended a pool party and spent much of the day swimming and reports that he has suffered from severe bouts OF BACK AND CHEST PAIN in the past
|
Sickle Cell Disease
-Swimming and lack of oxygen prompted sickling of HbS -Back and Extremity pain are due to VASO-OCCLUSIVE crises due to microvasculature blockage by sickled cells |
|
|
What is usually the first clinical manifestation of Sickle Cell Disease?
|
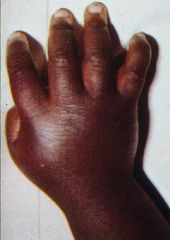
Dactylitis = infarctions in the bones of the digits
= hand-foot syndrome (swelling) |
|
|
Autosplenecomy
Howell-Jolly bodies |
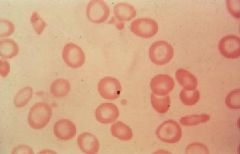
This is a blood smear from a patient with Sickle Cell Disease. What does it indicate? What is the cell called?
|
|
|
B is correct
-Decreased Hemoglobin -Increased HbA2 -usually asymptomatic |

-
|